")
")
| Issue |
Med Sci (Paris)
Volume 25, Number 12, Décembre 2009
Anticorps monoclonaux en thérapeutique
|
|
|---|---|---|
| Page(s) | 1078 - 1084 | |
| Section | I - De la conception à la production | |
| DOI | https://doi.org/10.1051/medsci/200925121078 | |
| Published online | 15 décembre 2009 | |
Du milligramme à la tonne d’anticorps monoclonaux
Outils et perspectives de production
Producing several hundred of kilograms of monoclonal antibodies for therapy: a constant challenge
1
Centre d’immunologie Pierre Fabre, 5, avenue Napoléon III, F74160 Saint-Julien-en-Genevois, France
2
Merck Research Laboratories, RY08Y-105, PO Box 2000, Rahway NJ 07065, États-Unis
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Identifier les anticorps monoclonaux à haut potentiel thérapeutique n’est pas une fin en soi. Pour se qualifier aux essais cliniques et potentiellement entrer sur un marché très convoité, le développement industriel des biomolécules utilise ses propres règles et technologies. Les pathologies à forte prévalence (oncologie, inflammation, etc.) peuvent nécessiter plusieurs centaines de kilogrammes d’anticorps par an. Il faut donc anticiper des investissements considérables en infrastructures bien avant toute autorisation de mise sur le marché. En parallèle, le choix du système d’expression, les équipements et les plateformes de production à mettre en oeuvre sont autant de considérations critiques auxquelles le développeur doit faire face. Dans un contexte compétitif et réglementé, l’enjeu est de réduire les délais et les coûts de développement, sans aucune contrepartie quant aux exigences de qualité attendues pour le produit final. Nous passerons en revue les principaux systèmes de production utilisés à ce jour et leurs évolutions espérées à court terme.
Abstract
Discovering and designing novel therapeutic monoclonal antibodies (mAb) is just the beginning. In order to support clinical evaluations and to reach the market place, rapid and cost effective production platforms are needed. Process development and production efficiency play a crucial role in this space since they influence the cost of good and ultimately wide access to these life-saving medications. Due to their therapeutic dosages and repeated uses, the yearly need for certain mAb, especially those used in the treatments of cancer and inflammation, amounts to several hundred of kilograms. Consequently, significant technological investments are needed to support these extraordinary large needs for such complex proteins, and the industry is constantly aiming at reducing production costs while maintaining product quality to high levels. This review discusses some of the critical scientific and engineering decisions, which span from the selection of cell-line expression platforms to choices of technologies, which influence mAbs cost of goods that need to be made along the development path of a therapeutic mAb.
© 2009 médecine/sciences - Inserm / SRMS
Les anticorps thérapeutiques constituent une classe de médicaments bien acceptée, tout particulièrement dans le domaine de l’oncologie, de l’inflammation et de la transplantation d’organes. À ce jour, plus de 30 anticorps ou fragments d’anticorps sont enregistrés et disponibles sur tout ou partie du marché mondial (→). Ces molécules représentent en 2008 un chiffre d’affaires dépassant les 20 milliards de dollars et les perspectives de croissance sont estimées à 14 % par an, loin devant la plupart des autres classes thérapeutiques [1, 2]. Ce succès devrait perdurer avec un triplement du nombre d’anticorps monoclonaux (Acm) qui sont entrés en phases cliniques au cours des dix dernières années. Les Acm cumulent deux avantages thérapeutiques : leur bonne tolérance physiologique et l’extraordinaire spécificité pour leur cible. Ces bénéfices sont toutefois mitigés par le coût des traitements lié à la complexité de développement et de production des Acm. En effet, il faudra 10 à 12 ans et environ 1 milliard de dollars pour passer de la découverte à la commercialisation d’un Acm [3].
(→) voir A. Beck et al., Tableau I, page 1026
La plupart des Acm commerciaux sont produits à partir de lignées cellulaires mammaliennes. Ces technologies requièrent un environnement de production complexe, des équipes hautement spécialisées et des matières premières onéreuses. Ces éléments expliquent en partie le coût des thérapies à base d’Acm. La capacité mondiale de production des Acm est aujourd’hui limitée si l’on considère le nombre de ces molécules en développement et leur chance de succès de mise sur le marché. Nous verrons toutefois que l’amélioration des productivités cellulaires vient tempérer cette limitation.
La construction de nouveaux sites de production commerciale est néanmoins une nécessité. Ces projets de plusieurs années requièrent anticipation, prise de risque et un budget d’au moins 500 millions de dollars [4]. Même si de nouveaux horizons s’ouvrent pour les applications thérapeutiques des Acm, il faut prévoir la multiplication de molécules couvrant des indications identiques ou similaires, et en conséquence, une diminution de la part de marché de chaque Acm.
La diminution des coûts globaux de développement et de production est devenue un enjeu majeur. Dans cet article, nous passerons en revue les méthodes actuelles de production des Acm et décrirons les innovations technologiques de développement et de production qui seront très probablement avantageuses économiquement dans les toutes prochaines années.
Les lignées cellulaires : des usines hautement spécialisées
Comme le détaillent A. Beck et al. (→) et R. Abès et al. (→) dans ce numéro, les anticorps sont des molécules dont la struc-ture primaire est complexe. Ils présentent aussi des motifs structuraux secondaires et tertiaires influençant largement leur activité. La synthèse d’Acm biologiquement actifs met en jeu des mécanismes biochimiques sophistiqués tels que le trafic dans différents compartiments cellulaires (→), les modifications post-traductionnelles et enfin la sécrétion. Le système de production et les conditions physicochimiques permettant la synthèse et la purification des Acm ont un impact majeur sur les rendements et la qualité de la molécule.
(→) page 1024
(→) page 1011
(→) voir S. Moutel et F. Perez, page 1173
Depuis la fin des années 1970 et jusqu’à la mise au point des méthodes récentes d’ingénierie cellulaire, seules certaines cellules mammaliennes ont été utilisées pour la production commerciale d’Acm. Dans un proche avenir, des systèmes alternatifs pourraient s’ajouter aux cellules mammaliennes pour la production d’Acm (voir ci-dessous).
Les hybridomes
Les tout premiers anticorps thérapeutiques ont été produits à partir de la technologie des hybridomes développée dans les années 1970 par Köhler et Milstein [5] (→). La découverte fut majeure et leur valut le prix Nobel en 1984. Les lymphocytes B (splénocytes) d’une souris immunisée avec l’antigène d’intérêt sont fusionnés avec des cellules immortalisées et non productrices d’anticorps. La culture in vitro de ce mélange cellulaire est réalisée en présence d’un système inhibiteur permettant uniquement la survie des cellules fusionnées ou hybridomes. Les clones (issus d’un hybridome unique produisant un seul type d’anticorps) peuvent être isolés et utilisés pour la production d’anticorps à grande échelle.
(→) voir J.L. Teillaud, page 1010 ; R. Abès et al., page 1011
Bien que les premiers anticorps recombinants aient été produits avec cette technologie, elle présente de sérieux inconvénients. Les Acm sont d’origine murine et leur injection répétée chez l’homme déclenche une réaction immunitaire avec formation d’anticorps dirigés contre ces anticorps de souris. Ce mécanisme de défense a pour conséquence l’élimination rapide des anticorps murins de l’organisme, réduisant ainsi sérieusement l’efficacité thérapeutique (→). Il a été démontré qu’environ 80 % des patients qui avaient reçu le premier anticorps murin commercial, OKT3, utilisé dans la prévention du rejet de greffe, développaient une réaction immunitaire [6]. Cette immunogénicité des anticorps murins chez l’homme explique que l’on ne trouve à ce jour que cinq Acm de souris sur le marché (→).
(→) voir P. Stas et I. Lasters, page 1070 ; G. Paintaud, page 1057
(→) voir A. Beck et al., Tableau I, page 1026
Les lignés cellulaires de mammifères
Des hybridomes murins aux anticorps humains
Les avancées de la biologie moléculaire ont permis de surpasser les limitations des hybridomes. Après sélection de l’hybridome d’intérêt, les séquences ADN codant l’anticorps sont isolées. Des approches graduelles ont été développées pour donner à l’anticorps de souris un aspect de plus en plus humain. Les anticorps chimériques sont construits à partir des régions constantes d’immunoglobulines humaines sur lesquelles sont attachées les régions variables de l’anticorps produit par l’hybridome correspondant. Par la suite, les techniques d’immunisation ont permis de construire des anticorps où seules les régions hypervariables de l’anticorps de souris sont greffées à la place des séquences équivalentes d’un anticorps humain (→).
(→) voir R. Abès et al., page 1011 ; M. Cogné et al., page 1149
Finalement, la production d’Acm complètement humains est devenue possible avec l’utilisation de souris transgéniques ou de banques de phages qui combinent la diversité des gènes d’immunoglobulines humaines. Les Acm Vectibix®, Humira® et Stelara® ont été obtenus par ces deux approches et sont 100 % humains (→).
(→) voir M. Cogné, page 1149 ; A. Beck et al.,Tableau I, page 1026
Après toutes ces manipulations qui, rappelons-le, interviennent au niveau du gène, la séquence d’ADN obtenue est clonée dans un vecteur d’expression et celui-ci est introduit dans une cellule mammalienne par transfection. L’intégration de l’ADN étranger dans le chromosome de la cellule est un prérequis pour l’expression future de l’Acm. Ce mécanisme se produit toutefois avec une fréquence relativement faible. Les cellules ayant intégré le vecteur sont généralement identifiées et isolées à l’aide d’un marqueur de sélection colocalisé sur le vecteur d’expression et qui code pour une séquence conférant aux cellules la capacité de proliférer en culture dans un milieu sélectif.
Production des Acm humanisés dans des systèmes cellulaires
De nombreuses lignées cellulaires immortalisées pourraient potentiellement être utilisées pour la production d’Acm. Dans les faits, à l’exception des deux Acm produits à partir d’hybridomes, tous les anticorps commerciaux sont produits en lignées mammaliennes de souris (NS0 et SP2/0, deux lignées de myélome murin) ou de hamster chinois (lignée CHO issue d’ovaire). Cette dernière a aujourd’hui la faveur des chercheurs industriels puisqu’elle est utilisée pour produire environ 50 % des Acm commerciaux (→) et des Acm en cours d’essais cliniques. Ces trois lignées peuvent être cultivées en suspension et s’accommodent assez bien du changement d’échelle (scale-up) des cultures, de l’étape in vitro au laboratoire jusqu’aux cultures en bioréacteur de grande capacité (20 000 l). La productivité cellulaire est l’un des critères essentiels du point de vue économique. Des progrès spectaculaires ont d’ailleurs été réalisés et les productivités multipliées par vingt ces dix dernières années. Actuellement, les productions industrielles sont de l’ordre de 1 à 4 g/l d’Acm et nous verrons plus loin, ce qui peut être envisagé pour dépasser ces seuils de productivité.
(→) voir A. Beck et al., Tableau I, page 1026
Le profil de glycosylation des IgG produites à partir des lignées NS0, SP2/0 ou CHO présente généralement de subtiles variations qui peu vent néanmoins avoir un impact important sur l’immunogénicité de la molécule (→). Ces glycoformes sont aujourd’hui parfaitement caractérisables et quantifiables. Par ailleurs, la maîtrise des procédés de production constamment évaluée par des contrôles rigoureux assure une constance dans le taux des glycoformes et un excellent profil de sécurité en clinique [6].
(→) voir P. Stas et I. Lasters, page 1070
Nous l’avons vu, les lignées cellulaires utilisées proviennent de rongeurs et les virus qu’elles intègrent ont été source d’inquiétudes. Au cours des quinze dernières années, des études approfondies ont été menées pour s’assurer de l’absence de virus pathogènes pour l’homme dans les cellules NS0, SP2/0 ou CHO. Néanmoins, des mesures spécifiques sont incluses dans les procédés de purification pour éliminer les particules virales avant l’utilisation des Acm en clinique.
Les systèmes émergents de production d’anticorps
Nouveaux modèles cellulaires
De nouveaux modèles cellulaires pourraient prochainement bousculer l’ordre établi avec les lignées de rongeurs. La lignée Per.C6® provient de cellules de rétine humaine immortalisées et ont un potentiel de culture à très haute densité. Plusieurs études ont démontré la capacité de Per.C6® de produire un Acm avec des rendements atteignant 10 g/l [7, 8]. Par nature, les protéines recombinantes issues de cette lignée ont un profil de glycosylation humain. Ces avantages sont des éléments déterminants pour permettre à la lignée Per.C6® de devenir une plateforme de production d’Acm.
D’autres lignées sont actuellement utilisées en développement : les cellules aviaires EBx® de la société Vivalis (→) et le myélome de rat YB2/0 des Laboratoires LFB (Laboratoire français du fractionnement et des biotechnologies) (→). Leur avantage principal est de produire des molécules avec un faible taux de fucose, ce qui en principe permet d’augmenter leur capacité de déclencher la cytotoxicité dépendante des anticorps (ADCC), une des fonctions effectrices des Acm.
(→) voir S. Olivier et M. Mehtali, page 1163
(→) voir R. Urbain et al., page 1141
Les bactéries, levures, champignons filamenteux, microalgues, plantes et animaux transgéniques sont autant de systèmes de production potentiellement utilisables pour la production d’anticorps [31, 32] (→). Les levures Pichia pastoris ont été modifiées génétiquement pour permettre l’expression de protéines avec des motifs humains de glycosylation. L’immunogénicité des biomolécules recombinantes est ainsi fortement réduite. Bien que la productivité de Pichia pastoris soit significativement inférieure à celle des cellules de mammifères décrites ci-dessus, des publications récentes décrivent des titres en protéines dépassant 2 g/l [9]. Ce système d’expression économiquement avantageux pourrait donc être utilisé dans un futur proche pour des productions industrielles.
(→) voir S. Olivier et M. Mehtali, page 1163
À la différence des lignées de rongeurs mentionnées plus haut, tous ces systèmes émergents qui ne sont pas encore enregistrés devront se confronter à une expertise réglementaire pointilleuse pour espérer se qualifier comme système de production.
Amélioration des lignées cellulaires par ingénierie génétique et métabolique
L’insertion du vecteur d’expression au niveau chromosomique est un phénomène aléatoire. Si l’intégration se produit dans une région hautement condensée de la chromatine, on observera généralement un faible taux d’expression de la protéine recombinante. L’environnement génomique du site d’intégration et la présence de modifications épigénétiques de l’ADN (méthylation) et des histones (acétylation, méthylation, phosphorylation, etc.) influencent considérablement l’efficacité de transcription du transgène. Les approches STAR (stabilizing and antirepressor), S/MAR (matrix associated regions1) ou UCOE (ubiquitous chromatin opening elements2), pour ne citer qu’elles, permettent l’ajout de séquences particulières d’ADN à proximité de l’insert codant l’Acm et induisent un environnement chromatinien favorable à la transcription [10]. Ces technologies nouvelles peuvent potentiellement simplifier la sélection de cellules ayant un haut rendement de production après transfection et également réduire les temps de développement [11].
L’ingénierie métabolique des cellules permet également d’améliorer ou de contrôler la qualité des Acm produits. La présence de motifs particuliers de glycosylation influence les interactions des Acm avec certaines cellules du système immunitaire. Ainsi, une lignée CHO dont le gène de la fucosyl transférase a été supprimé a permis l’expression d’Ac non fucosylés qui déclenchent une activité ADCC multipliée par 100. D’autre part, des modifications de la région Fc de l’Acm ont été introduites qui augmentent l’affinité de cette région vis-à-vis de son récepteur, améliorant la stabilité de l’anticorps in vivo [12–14].
Des modifications cellulaires ont été également décrites pour prolonger la durée de culture des cellules en bioréacteur. Une des stratégies se base par exemple sur l’utilisation d’ARN interférents pour inhiber des gènes proapoptotiques tels que Bax, Bak ou caspase 3 [15, 16]. À l’inverse, des copies additionnelles de gènes anti-apoptotiques tels que Bcl-2 et Bcl-XL ont été insérées dans la cellule productrice, augmentant sa survie, et donc entraînant l’augmentation des rendements de production [17]. Dans le cas d’une lignée surexprimant la protéine Bcl-XL, une augmentation de 80 % de la productivité volumétrique d’un anticorps humanisé a été démontrée par comparaison à celle de la cellule parentale [18].
L’amélioration de l’activité transcriptionnelle induite par ces modifications peut avoir des conséquences sur la capacité cellulaire d’assemblage des polypeptides tels que les anticorps. S’ils sont incorrectement ou incomplètement repliés, ils seront sécrétés en moins grande quantité et leur structure sera hétérogène. À ce stade, les protéines chaperonnes jouent un rôle essentiel pour assurer l’assemblage, le trafic cellulaire et la sécrétion des anticorps. Plusieurs travaux décrivent comment il est possible de modifier cette machinerie cellulaire, en jouant par exemple sur les niveaux relatifs d’expression des protéines BiP (immunoglobulin heavy chain binding protein) et PDI (protein disulfide isomerase) [19, 20].
De très nombreuses autres démarches ont été documentées dont l’ambition est d’améliorer le potentiel des lignées cellulaires pour la production d’anticorps recombinants (voir la revue de Barnes [10]). Même si ces principes généraux sont pour beaucoup au stade expérimental, il y a fort à parier que certaines approches se développeront jusqu’à être appliquées au stade industriel dans les prochaines années.
De la recherche à la commercialisation : changement d’échelle et de qualité
Les outils de production
Chaque stade impliquant l’utilisation d’Acm, de la recherche à la production industrielle, met en jeu des quantités et une qualité d’anticorps notablement différentes. Les exigences de pureté et de caractérisation sont évidemment croissantes et maximales lors de l’utilisation chez l’homme. Alors que quelques milligrammes suffisent pour des études au stade recherche, il n’est pas rare de produire plusieurs centaines de Kg du même anticorps destiné à certaines indications oncologiques (Figure 1).
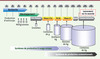 |
Figure 1. Représentation schématique des grandes étapes allant de la recherche à la production commerciale d’anticorps. La durée approximative des différentes phases du processus est indiquée ainsi que les quantités d’anticorps requises (cas des anticorps utilisés en oncologie). Les technologies à usage unique sont utilisables aujourd’hui pour des productions de l’ordre de quelques kilogrammes avec des perspectives dépassant la dizaine de kilogrammes à court terme. Au-delà, les sytèmes conventionnels de production (bioréacteurs de plusieurs milliers de litres) sont requis. |
Au niveau des procédés de préparation des protéines recombinantes, on distingue généralement les plates-formes upstream et downstream, respectivement dédiées aux activités de culture cellulaire et de purification [21].
La plate-forme upstream
Elle comprend toutes les étapes qui vont de la mise en culture du clone producteur de l’Acm jusqu’à la séparation des cellules et du milieu de culture produits par le dernier bioréacteur. Par extension, les étapes préliminaires incluant la génération de la lignée cellulaire (après transfection avec le vecteur d’intérêt), la sélection du clone et la réalisation de la banque cellulaire peuvent être inclues dans la plateforme upstream.
La sélection d’un clone, l’optimisation des conditions de culture et le changement d’échelle (de quelques millilitres à plusieurs milliers de litres) requièrent un véritable savoir-faire. Avec l’expertise et les cycles d’optimisation durant les phases de développement, il n’est pas rare d’obtenir des productivités cellulaires 5 à 10 fois supérieures à celle du clone initial. L’avantage économique est alors indéniable pour la mise en œuvre du procédé au stade industriel. Quel que soit le degré d’optimisation, l’enjeu, avant de passer à l’industrialisation, sera de démontrer la maîtrise du procédé tout en assurant la qualité finale du produit.
Les technologies de production ont notablement évolué ces dernières années, même si le principe de culture en bioréacteur reste le même. L’émergence des systèmes à usage unique a grandement facilité le travail des développeurs et producteurs [22, 23]. Il est aujourd’hui concevable de réaliser en système à usage unique toutes les étapes de culture cellulaire depuis la décongélation du tube de cellules de la banque cellulaire jusqu’au bioréacteur de 1 000 ou 2 000 litres. Ces bioréacteurs sont équipés d’une poche plastique stérile, d’un système d’agitation et de toutes les « connectiques » pour le contrôle de la culture cellulaire et les transferts de milieux. En fin de culture, les cellules sont récupérées, la poche est remplacée par une neuve, permettant au bioréacteur d’être de nouveau rapidement opérationnel. La plupart des équipements périphériques du bioréacteur (préparation des milieux, connectiques, filtration, etc.) sont également conçus sous une forme à usage unique. L’absence de stérilisation, de nettoyage et de contamination croisée sont les atouts principaux de ces systèmes. La limite de capacité actuelle des bioréacteurs (2 000 L) est compatible avec la production de lots d’anticorps destinés aux essais cliniques ou pour des petits marchés thérapeutiques (marchés de niche, nécessitant une production n’excédant pas quelques Kg). Dans le cadre de la production commerciale d’anticorps destinés au domaine oncologique par exemple, l’utilisation de bioréacteurs de conception plus traditionnelle et de grande capacité reste requise (jusqu’à 20 000 litres). Le lecteur pourra se référer à une revue récente traitant en profondeur les spécificités de la plateforme upstream [24].
La plate-forme downstream
Elle comprend l’ensemble des étapes permettant d’obtenir un anticorps purifié à partir du milieu de culture clarifié (débarrassé des cellules). Le principe actif obtenu devra ensuite être traité selon une formulation appropriée avant d’être flaconné pour les essais cliniques ou la commercialisation. Dans le cas des anticorps, les procédés standard de purification incluent le plus souvent une étape de chromatographie d’affinité (sur protéine A) suivie de deux étapes de chromatographie par échange d’ions ou par interaction hydrophobe. L’ensemble des méthodes mises en œuvre concourt à l’élimination des contaminants cellulaires ou apportés par le procédé (→). En particulier, les protéines contaminantes de la cellule hôte, l’ADN, la protéine A ou les virus potentiellement apportés par la lignée cellulaire doivent être éliminés ou limités à des concentrations inférieures aux normes réglementaires. Dans le cas des virus, l’élimination ou l’inactivation par au moins deux étapes orthogonales dans le procédé est un requis réglementaire. L’objectif de pureté s’accompagne d’un objectif de rendement optimal de la plateforme. Selon les choix faits par le développeur et en fonction des caractéristiques intrinsèques de l’anticorps, les rendements de purification généralement rencontrés sont de 60 à 70 %.
(→) voir L. Manache et al., page 1063
Avec l’amélioration spectaculaire des productivités cellulaires, la purification est devenue le goulet d’étranglement dans la production des anticorps [25]. Les étapes downstream représentent 60 à 70 % du coût global de production des anticorps. Les efforts de simplification de cette plateforme et de réduction des coûts sont devenus des enjeux majeurs. La mise au point d’alternatives à la protéine A [26], la création de nouvelles résines ou de nouvelles membranes de filtration chargées apporteront des solutions compétitives. De même, l’émergence des technologies à usage unique dans la plateforme downstream (filtration, préparation des tampons, connectiques, etc.) permet de réduire les temps du procédé tout en assurant la fiabilité de la qualité du principe actif. La mise à disposition rapide d’anticorps pour des essais cliniques est absolument essentielle dans le cycle de développement et la perspective de succès commercial [27, 28].
En parallèle des plateformes upstream et downstream, des méthodes analytiques sont mises en place pour caractériser l’anticorps d’intérêt. Compte tenu de l’exigence croissante de pureté et de caractérisation, on dispose aujourd’hui de techniques analytiques repoussant toujours plus loin les limites de quantification des subtiles variations observées dans la structure des anticorps [29] (→).
(→) voir L. Manache et al., page 1063
Conclusion
Comme beaucoup d’articles de ce numéro en témoignent, les anticorps sont des molécules majeures sur le plan thérapeutique. De nombreuses entités pharmaceutiques se sont engouffrées dans ce créneau porteur. La compétition est donc active, d’autant plus que les cibles thérapeutiques visées sont bien moins nombreuses que les centaines d’anticorps en développement. D’un autre côté, les coûts élevés de la recherche, du développement et de la production des anticorps en ont fait des thérapies onéreuses. Les politiques de remboursement revues à la baisse et l’émergence des biosimilaires mettent l’industrie pharmaceutique face à un défi nouveau. Les risques financiers et l’effervescence de la compétition stimulent les esprits à la recherche de technologies plus rapides, plus fiables et plus économiques [30].
Les productivités cellulaires ont littéralement explosé ces dix dernières années. Ce paramètre répond en partie aux aspects économiques parce que simplement il permet de diminuer les volumes des bioréacteurs requis pour obtenir une quantité égale d’anticorps. Si l’avantage upstream est évident, tout le monde s’accorde aujourd’hui sur la nécessité de trouver des solutions nouvelles à la plateforme downstream. C’est l’un des défis immédiats que l’industrie biopharmaceutique devra relever.
Mais dès à présent, avec le nombre d’anticorps en développement et les systèmes technologiques en place, cette même industrie offre des perspectives optimistes pour apporter aux patients de nouvelles biomolécules thérapeutiques.
Conflit D’Intérêts
O. Cochet déclare avoir des liens durables avec l’entreprise Institut de Recherche Pierre Fabre.
MAR : éléments d’ADN qui se lient à la matrice nucléaire et contribuent à créer une organisation supérieure de la chromatine au sein de boucles indépendantes où l’expression des gènes est coordonnée.
Il peut s’agir par exemple de régions promotrices de gènes de ménage exprimés de façon ubiquitaire et à un fort taux.
Références
- Reichert JM. Monoclonal antibodies as innovative therapeutics. Curr Pharm Biotechnol 2008; 9 : 423–30. [Google Scholar]
- Scolnik P. mAbs: a business perspective. mAbs 2009; 1 : 1–6. [Google Scholar]
- Reichert J. Number of mAbs entering clinical study nearly tripled in last decade. Tufts Center for Study of Drug Development Impact Report 2008; 10 :1–4. [Google Scholar]
- Werner R. Economic aspects of commercial manufacture of biopharmaceuticals. J Biotechnol 2004; 113 : 171–82. [Google Scholar]
- Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975; 256 : 495–7. [Google Scholar]
- Roskos L, Davis C, Schwab G. The clinical pharmacology of therapeutic monoclonal antibodies. Drug Dev Res 2004; 61 : 108–20. [Google Scholar]
- Jones D, Kroos N, Anema R, et al. High-level expression of recombinant IgG in the human cell line per.c6. Biotechnol Prog 2003; 19 : 163–8. [Google Scholar]
- Yallop C, Raamsman M, Zuijderwijk M, et al. High level production of recombinant IgG in the human cell line PER.C6. In : Francesc Gòdia MF, ed. Animal cell technology meets genomics. Amsterdam : Springer Netherlands, 2005 : 533–6. [Google Scholar]
- Gurramkonda C, Adnan A, Gabel T, et al. Simple high-cell density fed-batch technique for high-level recombinant protein production with Pichia pastoris: application to intracellular production of Hepatitis B surface antigen. Microb Cell Fact 2009; 8 :13. [Google Scholar]
- Barnes LM, Dickson AJ. Mammalian cell factories for efficient and stable protein expression. Curr Opin Biotechnol 2006; 17 : 381–6. [Google Scholar]
- Kwaks TH, Otte AP. Employing epigenetics to augment the expression of therapeutic proteins in mammalian cells. Trends Biotechnol 2006; 24 : 137–42. [Google Scholar]
- Carter PJ. Potent antibody therapeutics by design. Nat Rev Immunol 2006; 6 : 343–57. [Google Scholar]
- Shields RL, Namenuk AK, Hong K, et al. High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J Biol Chem 2001; 276 : 6591–604. [Google Scholar]
- Vaccaro C, Zhou J, Ober RJ, et al. Engineering the Fc region of immunoglobulin G to modulate in vivo antibody levels. Nat Biotechnol 2005; 23 : 1283–8. [Google Scholar]
- Lim SF, Chuan KH, Liu S, et al. RNAi suppression of Bax and Bak enhances viability in fed-batch cultures of CHO cells. Metab Eng 2006; 8 : 509–22. [Google Scholar]
- Sung YH, Hwang SJ, Lee GM. Influence of down-regulation of caspase-3 by siRNAs on sodium-butyrate-induced apoptotic cell death of Chinese hamster ovary cells producing thrombopoietin. Metab Eng 2005; 7 : 457–66. [Google Scholar]
- Figueroa B Jr, Sauerwald TM, Mastrangelo AJ, et al. Comparison of Bcl-2 to a Bcl-2 deletion mutant for mammalian cells exposed to culture insults. Biotechnol Bioeng 2001; 73 : 211–22. [Google Scholar]
- Chiang GG, Sisk WP. Bcl-x(L) mediates increased production of humanized monoclonal antibodies in Chinese hamster ovary cells. Biotechnol Bioeng 2005; 91 : 779–92. [Google Scholar]
- Borth N, Mattanovich D, Kunert R, et al. Effect of increased expression of protein disulfide isomerase and heavy chain binding protein on antibody secretion in a recombinant CHO cell line. Biotechnol Prog 2005; 21 : 106–11. [Google Scholar]
- Davis R, Schooley K, Rasmussen B, et al. Effect of PDI overexpression on recombinant protein secretion in CHO cells. Biotechnol Prog 2000; 16 : 736–43. [Google Scholar]
- Bergemann K, Eckermann C, Garidel P, et al. Production and downstream processing. In : Dübel S, ed. Handbook of therapeutic antibodies. New York : Wiley, 2007 : 199–237. [Google Scholar]
- Foulon A, Trach F, Pralong A, et al. Using disposables in an antibody production process. BioProcess Int 2008; 6 : 12–7. [Google Scholar]
- The single-use technology guidebook. BioProcess Int 2008; 6 (suppl 3). [Google Scholar]
- Chartrain M, Chu L. Development and production of commercial therapeutic monoclonal antibodies in mammalian cell expression systems: an overview of the current upstream technologies. Curr Pharm Biotechnol 2008; 9 : 447–67. [Google Scholar]
- Scott C. Proactive debottlenecking. Planning ahead for the downstream bottleneck. BioProcess Int 2008; 6 : 32–42. [Google Scholar]
- Arunakumari A, Wang J, Figueroa B Jr. Advances in non-protein A purification processes for human monoclonal antibodies. Biopharm Int 2009 (suppl mars). [Google Scholar]
- Seaman C, Fries S, Beck A, et al. Cell cultivation process transfer and scale-up. In support of production of early clinical supplies of an anti IGF-1R antibody (part 1). BioProcess Int 2008; 6 : 26–36. [Google Scholar]
- Seaman C, Fries S, Beck A, et al. Cell cultivation process transfer and scale-up. In support of production of early clinical supplies of an anti IGF-1R antibody (part 2). BioProcess Int 2008; 6 : 34–42. [Google Scholar]
- Beck A, Wagner-Rousset E, Bussat MC, et al. Trends in glycosylation, glycoanalysis and glycoengineering of therapeutic antibodies and Fc-fusion proteins. Curr Pharm Biotechnol 2008; 9 : 482–501. [Google Scholar]
- Farid SS. Process economics of industrial monoclonal antibody manufacture. J Chromatogr B Analyt Technol Biomed Life Sci 2007; 848 : 8–18. [Google Scholar]
- Faye L, Champey Y. Plants, medicine and genetics, which applications for tomorrow ? Med Sci (Paris) 2008; 24 : 939–45. [Google Scholar]
- Cadoret JP, Bardor M, Lerouge P, et al. Microalgae as cell factories producing recombinant commercial proteins. Med Sci (Paris) 2008; 24 : 375–82. [Google Scholar]
Liste des figures
|
Figure 1. Représentation schématique des grandes étapes allant de la recherche à la production commerciale d’anticorps. La durée approximative des différentes phases du processus est indiquée ainsi que les quantités d’anticorps requises (cas des anticorps utilisés en oncologie). Les technologies à usage unique sont utilisables aujourd’hui pour des productions de l’ordre de quelques kilogrammes avec des perspectives dépassant la dizaine de kilogrammes à court terme. Au-delà, les sytèmes conventionnels de production (bioréacteurs de plusieurs milliers de litres) sont requis. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.