")
")
| Issue |
Med Sci (Paris)
Volume 25, Number 6-7, Juin-Juillet 2009
|
|
|---|---|---|
| Page(s) | 608 - 616 | |
| Section | Dossier Biologie des systèmes | |
| DOI | https://doi.org/10.1051/medsci/2009256-7608 | |
| Published online | 15 June 2009 | |
Modélisation intégrative prédictive et biologie expérimentale
Un processus synergique remarquablement efficace au service de la recherche médicale
Production and implementation of predictive biological models
Bio-Modeling Systems, 26, rue Saint-Lambert, 75015 Paris, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
La nécessité d’élaborer et mettre en oeuvre des méthodes permettant une modélisation prédictive cohérente, fiable et précise des systèmes biologiques est devenue une évidence tant les enjeux économiques qui y sont liés sont importants. Cela est vrai au niveau industriel (pharmacologie, agroalimentaire et environnement, etc.) comme au niveau public (santé, recherche, etc.). On admet communément que la modélisation est avant tout une affaire d’informatique. Bien que cela se vérifie dans le cas des systèmes « continus » ou peu complexes, il n’en est pas de même en ce qui concerne les systèmes « discontinus » ou hyper-complexes que sont les systèmes vivants par exemple. Dans ces domaines, la modélisation devient une affaire de biologie assistée par l’informatique, et non pas le contraire. Nous tenterons dans cet article d’en développer les raisons et nous l’illustrerons à l’aide d’exemples concrets.
Abstract
The production and implementation of methodologies allowing the construction of coherent, precise and trustworthy predictive biological models has become an inescapable necessity. The financial stakes attached to this reality are very high indeed, be it in the public or the private domains (health, research, pharma and foodstuffs industries, environment, etc.). Modelling biological systems is widely presented as a problem in computational sciences. While certainly very true for low complexity, practically continuous systems, this view cannot be upheld in the case of discontinuous, hyper-complex systems such as living entities. In these domains, modelling becomes a problem in biology assisted by computational sciences and certainly not the obverse. The following article will attempt to demonstrate why it is so, using concrete examples.
© 2009 médecine/sciences - Inserm / SRMS
Le développement d’un médicament est principalement un problème d’intégration d’informations et de gestion de connaissances. Connaître les cibles potentielles d’une molécule et les fonctions de ces cibles est une chose. Connaître les mécanismes physiologiques qui doivent être ciblés et la façon dont ils doivent être manipulés pour obtenir un impact thérapeutique donné en est une autre. Or, non seulement ces deux aspects ne sont pas interchangeables, mais le succès du développement d’un médicament résulte principalement de la manipulation cohérente d’un système physiologique dans son ensemble et non de la manipulation d’une cible dans un contexte restreint.
Cela impose une nouvelle approche qui requiert en tout premier lieu la modélisation des mécanismes physiopathologiques concernés ; elle sera suivie de vérifications expérimentales in vivo dont l’objectif est de valider et/ou d’infirmer les points clés qui sous-tendent le modèle. Ces résultats expérimentaux sont ensuite intégrés dans le modèle pour en corriger les erreurs, aboutissant à un modèle qui décrit en détail les mécanismes associés à l’instauration puis au développement d’une pathologie donnée et prédit les moyens qui devront être mis en œuvre pour prévenir, ou traiter, cette maladie.
Qu’est-ce qu’un « modèle » et que peut-on en attendre ?
Pour atteindre ce but, les va-et-vient entre modélisations et vérifications expérimentales sont incontournables. En effet, un modèle ne peut être autre chose qu’une approximation de la réalité. Plus le phénomène biologique devant être modélisé est complexe, plus l’approximation que constitue le modèle est grossière. Il est donc indispensable que ce modèle soit confronté à la réalité biologique qu’il est censé représenter.
Cette démarche est à l’origine d’un processus synergique d’une efficacité tout à fait étonnante : un modèle indique très précisément ce qui devrait être observé expérimentalement, quand, où, comment et pourquoi. De ce fait, les résultats expérimentaux ne peuvent plus être sujets à conjectures et les erreurs du modèle sont très vite décelées et corrigées. Mais il y a beaucoup plus. Les expériences destinées à tester un modèle révèlent souvent des résultats totalement inattendus qui, intégrés au modèle, permettent des avancées d’une rapidité et d’une pertinence remarquables. Toutefois, pour obtenir les connaissances fonctionnelles requises, il est nécessaire d’intégrer une énorme quantité d’informations, très largement disséminées sous une multitude de formes et de représentations, et correspondant à une immense complexité.
La vision « classique » du processus de modélisation
La valeur de l’information
La masse d’informations biologiques produite au cours de ces cinquante dernières années est d’une telle amplitude que la littérature scientifique s’est complexifiée au-delà de toute mesure.
Dans la façon « classique » de traiter le problème de la modélisation, il est indispensable de construire des bases de données « informationnelles » les plus exhaustives possible. Il est également vital de pouvoir exploiter le contenu de ces bases de données avec la plus grande flexibilité possible [1–5]. On sous-entend par là que, d’un point de vue analytique, toute « information » est potentiellement vitale, et que toute information est exploitable et doit être exploitée. Il en résulte des stratégies de plus en plus complexes de fouille de texte, d’archivage, de mise en relation, de mise à jour et d’exploitation.
Ces approches impliquent donc un processus de sélection positive : « l’information » est traitée comme une entité « fiable » à laquelle une valeur nominale définie (positive ou négative) peut être assignée.
Or, les faits démontrent que la valeur intrinsèque d’une information n’est que relative et que cette valeur peut être profondément modifiée tant par les contextes auxquels elle peut être associée que par d’autres informations qui ne lui sont pas directement liées. De plus, une grande partie de cette information résulte de l’adoption d’approches réductionnistes qui, pour pallier les difficultés souvent insurmontables des systèmes in vivo, ont fait massivement appel à des expérimentations in vitro, qui ne concernent que des aspects très restreints du phénomène biologique étudié ; de plus, ces approches in vitro se font dans des conditions très hétérogènes, à partir de sources très diverses de matériel biologique et dans des conditions très éloignées de la réalité physiologique in vivo.
De fait, l’avalanche d’informations qui en résulte a trois caractéristiques qui ne peuvent être contrôlées ni par ses producteurs, ni par ses utilisateurs potentiels. Toute information ainsi produite est toujours (1) incomplète, mais nul ne sait jusqu’à quel point ; (2) biaisée, mais nul ne sait ni comment ni jusqu’à quel point, et (3) erronée, mais nul ne sait jusqu’à quel point.
Il en résulte des conséquences majeures sur toute approche analytique basée sur la sélection positive. Non seulement tout ce que désigne le terme d’« information » n’est pas nécessairement utile et/ou utilisable mais, de plus, la valeur analytique des bases de données s’en trouve bouleversée. Le contenu de ces bases est très largement entaché par une accumulation d’incohérences. Le « vrai » se trouve mélangé avec « l’incertain » sans qu’il soit possible d’éliminer le « faux » et encore moins de déterminer dans quels contextes le « vrai » peut tout à coup devenir « faux ». En outre, plus la complexité dans laquelle s’insère « l’information » est grande, plus les effets négatifs qu’auront ces inconsistances sur tout processus analytique seront importants.
Conséquences sur la modélisation des imperfections des bases de données
Ces difficultés sont à l’origine des problèmes rencontrés par les diverses tentatives de modélisation à grande échelle des systèmes vivants basées sur l’exploitation des contenus de bases de données « informationnelles » associée à des approches analytiques largement mathématiques [5, 6]. Une approche mathématique requiert soit l’identification des variables devant être prises en compte dans le processus de modélisation, soit une quasi-linéarité fonctionnelle du système que l’on souhaite modéliser. En effet, nous ne savons toujours pas résoudre les équations différentielles non-linéaires : les approches de choix restent, soit de prendre en compte une connaissance au moins qualitative des variables (ce qui permet de résoudre les problèmes de non linéarité), soit d’introduire des constantes en nombre suffisant pour approcher la linéarité.
Or, le vivant est un exemple type de « système hypercomplexe » et les faits démontrent que non seulement nos connaissances, en termes de variables impliquées, sont extrêmement limitées, mais la linéarité fonctionnelle n’y est que très rarement présente. Alors que le génome humain contient quelques 25 000 gènes, le transcriptome, lui, contient au moins 2 x 105 entités et le protéome plus d’un million de composantes simples. De plus, ces très nombreuses composantes établissent en permanence des interactions dynamiques entre elles, sont sujettes à de multiples modifications structurelles qui ont des conséquences fonctionnelles et donnent naissance à des structures complexes plus ou moins pérennes, capables d’auto-organisation sur plusieurs niveaux d’échelles et soumises tant aux lois du chaos qu’à celles de l’évolution. Non seulement ces structures entraînent l’apparition de comportements et de hiérarchies qui ne peuvent pas être déduits des propriétés des composantes, mais elles sont majoritairement pluri-fonctionnelles et chaque fonction est largement dépendante du contexte [24]. Nous sommes donc en présence d’un système intégratif et non linéaire.
Toute approche tendant à la linéarité par approximation ne peut donc qu’engendrer des artifices, à plus forte raison si elle implique l’exploitation par sélection positive de masses d’information dont la validité physiologique est douteuse.
Comment, dans ces conditions, aborder le problème de la modélisation biologique prédictive ?
Une approche « événementielle et relativiste »
L’approche et les méthodologies que nous avons développées (CADI) se fondent sur une interprétation radicalement nouvelle du problème et elles ont fait leurs preuves dans des domaines aussi variés que la cancérologie, les maladies neuro-dégénératives et l’embryologie [7–12].
Les fonctions des composantes biologiques sont dépendantes du contexte [2–6]. Dans les systèmes vivants, les événements dictent aux contextes comment ils doivent se déformer, les contextes dictent aux composantes comment elles doivent se comporter et les composantes dictent aux événements comment ils doivent naître.
Cette règle de fonctionnement « en boucle » entraîne un changement majeur dans l’utilisation de « l’information ». Un traitement en termes de composantes et de fonctions n’a ici plus lieu d’être. Il devient nécessaire d’adopter une approche événementielle et relativiste (inconséquences, poids relatifs, entropie, etc.), qui permette d’utiliser le caractère incomplet, biaisé, et en partie erroné de toute information pour ensuite mettre en œuvre un système analytique basé sur la sélection, négative cette fois, d’hypothèses construites à partir des relations apparentes entre « informations ».
Il en résulte que, toute information devant être considérée comme a priori suspecte, l’utilisation des bases de données s’en trouve radicalement modifiée.
Non seulement il n’est plus nécessaire qu’une base de données « informationnelle » soit exhaustive, mais son contenu n’a plus aucune importance analytique. L’index qui lui est associé devient alors très important (Figure 1). Non pas parce que cet index serait crédible, il ne l’est pas plus que le contenu de la base de données, mais parce qu’il va permettre de formuler très rapidement de nombreuses hypothèses que l’on va systématiquement chercher à détruire en utilisant les multiples recoupements que permettent les redondances dans l’ensemble de la littérature scientifique. Rien n’est plus facile à détruire qu’une hypothèse erronée et les éléments qui permettent de la détruire peuvent être utilisés pour suggérer une nouvelle hypothèse un peu plus solide que la précédente, et ainsi de suite jusqu’à l’obtention d’une hypothèse qui ne peut plus être réfutée (Figure 1) parce qu’elle est confortée par de nombreux recoupements. Cela ne signifie pas pour autant que cette hypothèse soit juste. Cela signifie simplement qu’elle doit être prise au sérieux.
 |
Figure 1. Approche événementielle et relativiste de l’information. A. Le processus d’intégration commence par la définition du problème biomédical devant être traité (cancer du sein dépendant de l’activation de l’oncogène ras, hypercholestérolémie familiale, détoxification d’une molécule déterminée, etc.), ce qui permet de définir des contextes physiopathologiques. B. Toutes les informations stockées dans les bases de données publiques ayant un lien quelconque avec ces contextes sont rapatriées et structurées sous forme de base de données indexée (en termes de composantes biologiques, d’interactions, d’interdépendances, etc.). Si des données expérimentales sont disponibles, elles sont injectées dans cette base de données et sont prises en compte dans une nouvelle indexation. C. Les arborescences de l’index sont ensuite visualisées et les éléments donnant naissance aux embranchements sont utilisés pour générer des hypothèses fonctionnelles. D. Chaque hypothèse ainsi générée est alors testée contre l’ensemble des bases de données publiques, le but recherché ici étant la destruction de l’hypothèse. E. Les éléments d’information permettant de la détruire sont alors utilisés, conjointement avec les éléments d’arborescence, pour générer une nouvelle hypothèse qui est à son tour soumise au processus de destruction et ainsi de suite, de façon itérative, jusqu’à obtention d’une hypothèse ne pouvant pas être détruite. F. Les hypothèses non détruites accumulées sont alors intégrées en méta-hypothèses qui sont à leur tour soumises au même processus de destruction. Ce procédé, qui est étonnamment rapide (moins d’un an pour une maladie complexe et largement incomprise telle que la maladie de Creutzfeldt-Jakob), amène à la construction d’un modèle biologique, souvent très détaillé et directement vérifiable par expérimentations indépendantes. |
Cette approche permet de reconstituer des événements qui définissent des déformations de contextes ; ceux-ci, à leur tour, impliquent des comportements et des fonctions définis en termes de composantes, ce qui entraîne l’interdiction d’événements définis et l’apparition d’autres événements définis, etc.
Il en résulte un modèle entièrement documenté par un ensemble important d’informations bien identifiées et par de multiples recoupements ; ce modèle suggère des mécanismes cohérents pour de nombreuses observations qui restaient jusqu’alors sans explication et décrit de façon très détaillée (1) les conditions qui permettent l’apparition d’une pathologie, (2) les mécanismes qui sous-tendent son développement et (3) les modes d’interventions par lesquels elle pourrait être prévenue ou enrayée.
Bien entendu, ce type de modèle reste entièrement théorique et doit impérativement être vérifié expérimentalement. Toutefois, il fait des prédictions fonctionnelles très précises qui peuvent facilement être vérifiées in vivo dans un modèle animal.
La modélisation intégrative prédictive en pratique
Prenons un exemple concret
Le canal de Müller est une structure embryonnaire présente chez les mammifères mâles et femelles. Chez ces dernières, il est à l’origine de l’utérus et des trompes de Fallope ; chez le mâle, en réponse à une hormone (AMH, antimullerian hormone), il est l’objet d’un processus de régression qui le transforme en un cordon fibreux compact. La régression qui se propage comme une vague débute à partir de l’extrémité postérieure du canal alors que son extrémité antérieure continue à se développer normalement. Ce canal est constitué d’un manchon épais de cellules mésenchymateuses (le mésenchyme est un tissu embryonnaire à partir duquel sont formés les vaisseaux, les muscles, le squelette et le cartilage) entourant un tube mince, bordé d’une seule couche de cellules épithéliales séparées des autres cellules par une fine membrane fibreuse. Alors que seules les cellules mésenchymateuses expriment le récepteur à l’AMH (et peuvent donc répondre à l’hormone), ce sont les cellules épithéliales qui, bien que n’exprimant pas le récepteur AMH et n’étant jamais directement exposées à l’hormone, entraînent le phénomène de régression en étant pour partie converties en cellules mésenchymateuses et pour partie poussées à la mort [13, 14]. Comment est-ce que tout cela fonctionne ?
Les étapes initiales de modélisation
Au cours des soixante dernières années [15], de nombreux travaux ont été entrepris dans ce domaine, sans apporter de réponse fonctionnelle à ce phénomène [16, 17].
Le travail de modélisation a été entrepris à partir d’observations expérimentales décrivant certains des mécanismes mis en œuvre durant le processus de régression (Figure 2). Mais pour comprendre ce processus, il était nécessaire de remonter à une période du développement antérieure à l’apparition de l’AMH dans le système, pour laquelle aucune donnée expérimentale directe n’était disponible. Cela permettait de définir les contextes fonctionnels dans lesquels le récepteur AMH allait apparaître, quels mécanismes étaient interdits par ces contextes, quelles déformations ceux-ci allaient subir, quelles en seraient les conséquences fonctionnelles en termes de composantes et quels nouveaux événements découleraient de ce processus. La démarche décrite plus haut a été appliquée à partir de l’index d’une base de données « embryologiques » et les résultats de cette modélisation sont représentés dans la Figure 3.
 |
Figure 2. Observations expérimentales d’origine associées au processus de régression du canal de Müller chez la souris. Ces données ont été obtenues par le Pr F. Xavier (CNRS et ECP) et son équipe. Deux voies principales sont activées dans les cellules mésenchymateuses en réponse à l’AMH. La voie PI3K, qui aboutit à une accumulation cytoplasmique massive de β-caténine, et la voie Smad, qui résulte de la présence de complexes composés des protéines Smad4, β-caténine et Lef dans le noyau de ces cellules. Les points d’interrogation signifient l’incertitude quant à la forme structurale du récepteur à l’AMH entraînant l’activation de ces voies de signalisation (le récepteur AMHRII seul ou sa forme complexe AMHRII-ALK). |
 |
Figure 3. Résultats simplifiés de modélisation décrivant les conditions requises pour l’expression d’AMHRII dans les cellules mésenchymateuses. Ce diagramme décrit les communications entre cellules épithéliales (peu nombreuses) et cellules mésenchymateuses ainsi que l’état physiologique de ces cellules au moment où la réponse à l’AMH va commencer. Les lignes rouges indiquent les mécanismes et les interconnexions qui se trouvent interdites dans ce contexte. |
Avec cette carte physiologique en main, il devenait possible de s’adresser aux mécanismes du processus de régression.
Les mécanismes de la régression müllerienne
Il s’avère que ce processus se déroule en trois étapes distinctes dans l’espace et dans le temps (Figure 4), en raison des mécanismes interdits par les contextes initiaux. Une des caractéristiques du récepteur de l’AMH est de fonctionner sous la forme de complexes hétérodimères composés de l’association d’une protéine appelée AMHRII (le récepteur auquel se lie l’AMH) et d’une autre protéine appelée ALK (anaplastic lymphoma kinase). Cette dernière est elle-même le récepteur d’un autre type d’hormone appelé BMP (bone morphogenetic protein). Pour induire une réponse à l’AMH, les cellules mésenchymateuses doivent donc, selon la vision classique, exprimer AMHRII et ALK. Mais elles doivent organiser la distribution de ces protéines à la surface cellulaire de façon à ce que lorsque l’AMH se lie au récepteur AMHRII, ce dernier provoque le recrutement d’ALK et forme ainsi le complexe activé final décrit dans la littérature. Or, toute une série de réactions qui sont inhibées lorsque AHMRII est exprimé (et dont l’explication dépasse le cadre de cet article) empêchent le recrutement de la protéine ALK. Donc, à l’extrémité postérieure du canal, où le récepteur AMHRII est exprimé en premier, ce récepteur fonctionne initialement seul.
Cela entraîne une série de réponses qui vont avoir plusieurs conséquences : (1) permettre partiellement le recrutement de ALK ; (2) préparer la dissociation des connexions entre les cellules mésenchymateuses et (3) induire, par un effet de diffusion, les conditions permettant l’expression de AMHRII dans les cellules en aval. Cela déclenche donc la vague de propagation (Figure 4A). Dès lors (seconde phase), le recrutement de ALK par AMHRII activé devient partiellement possible et la réponse à l’AMH est transmise aussi bien par AMHRII que par le complexe AMHRII-ALK. Il en résulte : (1) la réactivation de certains des mécanismes initialement bloqués (voies et interactions représentées par les lignes rouges dans la Figure 3) ; (2) la dissociation locale des cellules du manchon et (3) toute une série de restructurations internes de ces cellules qui changent de forme (Figure 4B) et vont maintenant transmettre la réponse à l’AMH principalement via les seuls complexes AMHRII-ALK. Cette étape en retour induit la troisième phase (Figure 4C) qui est caractérisée par la mise en œuvre d’une série d’événements sous l’effet, en partie, de la réactivation des mécanismes initialement bloqués et en partie de leurs conséquences indirectes. Il en résulte la sécrétion locale d’enzymes qui détruisent partiellement la membrane fibreuse séparant les cellules épithéliales des cellules mésenchymateuses. En réponse à cette destruction partielle, les cellules épithéliales encore attachées à des fragments intacts de membrane fibreuse subissent une transformation appelée transition epithéliale-mésenchymateuse (EMT, phénomène souvent rencontré en oncologie) qui les convertit en cellules mésenchymateuses ; celles qui ne sont plus en contact qu’avec des débris de cette membrane se suicident (apoptose), entraînant la formation locale d’un cordon compact, constitué uniquement de cellules mésenchymateuses insérées dans un maillage fibreux (fibronectine). Pendant ce temps, la vague de régression initiée plus haut continue à se propager de proche en proche le long du canal à une vitesse bien supérieure à celle de la croissance de cette structure.
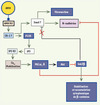 |
Figure 4A Résultats simplifiés de modélisation décrivant la première phase de réponse à l’AMH dans les cellules mésenchymateuses. Ici, seul le récepteur AMHRII est actif (imposé par des effets de fluidique membranaire). Le signal résultant est l’activation de certains éléments de la voie PI3K, aboutissant à l’accumulation d’une fraction de la β-caténine cellulaire et la sécrétion de fibronectine, préparant la rupture des contacts entre cellules mésenchymateuses. Ces événements concernent principalement les cellules à la partie superficielle des contacts avec le système vasculaire dans le manchon (voir texte), les cellules des couches internes n’ayant pas encore un accès libre à l’hormone. Les lignes et croix rouges indiquent des voies devenues interdites. Les points d’interrogation indiquent l’incertitude quant à l’identité exacte de la composante correspondante. Les boîtes bleues correspondent aux éléments vérifiés in vivo chez la souris par F. Xavier et son équipe au CNRS. |
 |
Figure 4B Résultats simplifiés de modélisation décrivant la seconde phase de réponse à l’AMH dans les cellules mésenchymateuses. Les complexes AMHRII-ALK sont maintenant activés alors que AMHRII seul reste encore fonctionnel. Il en résulte la mobilisation et la stabilisation de la β-caténine membranaire, entraînant la décomposition des contacts entre cellules mésenchymateuses, et la ré-induction partielle des voies Smad qui étaient jusqu’alors inhibées. À ce stade, les cellules mésenchymateuses superficielles se dissocient et changent de forme (flèches annotées « 2 »). Les cellules des couches internes du manchon sont maintenant exposées à l’AMH et entrent en phase 1 (flèches annotées « 1 »). Les boîtes entourées d’un cadre gras correspondent aux éléments vérifiés in vivo chez la souris par F. Xavier et son équipe au CNRS. |
 |
Figure 4C Résultats simplifiés de modélisation décrivant la troisième phase de réponse à l’AMH dans les cellules mésenchymateuses. En réponse à la décomposition des contacts entre cellules (phase 2), toutes les cellules mésenchymateuses, sur la profondeur du manchon, sont localement exposées à l’AMH et les complexes AMHRII-ALK sont maintenant les récepteurs majoritairement actifs. Cela entraîne la réactivation complète des voies Smad, la sécrétion d’enzymes (MMP3 et 9) et la dégradation partielle de la membrane fibreuse séparant les cellules épithéliales des cellules mésenchymateuses (flèches annotées 3 et 4). Ces dernières sont protégées des effets de ces enzymes par la fibronectine sécrétée durant les phases 1 et 2. En retour, la dégradation partielle de la membrane fibreuse induit (1) la mort des cellules épithéliales ancrées à des fibres dégradées et (2) la conversion en cellules mésenchymateuses de celles qui sont ancrées à des régions de la membrane encore intactes. Les croix rouges indiquent des interactions devenues interdites. L’orientation des flèches indique des gènes dont l’expression est fortement augmentée (pointe vers le haut) et fortement inhibée (pointe vers le bas). Les boîtes entourées d’un cadre gras correspondent aux éléments vérifiés in vivo chez la souris par le F. Xavier et son équipe au CNRS. |
Les événements clés décrits ci-dessus et dans les Figures 4A, B et C ont été expérimentalement vérifiés in vivo chez la souris (publication scientifique en cours).
Les modèles biologiques intégrés et leurs applications
Ce type de modèle intégré n’est en aucune façon l’équivalent d’une « cellule virtuelle ». Bien que décrivant des enchaînements de mécanismes intracellulaires associés à un état physiologique défini, ces modèles intègrent aussi des événements touchant des types cellulaires ou tissulaires différents, affectés par ce changement d’état (hypercholestérolémie familiale, maladie de Creutzfeld-Jakob, etc.). Ce type de modèle est donc à la fois : (1) une représentation dynamique dans le temps des enchaînements de mécanismes intracellulaires associés au développement et à l’instauration d’un changement d’état, et (2) une image des modifications physiologiques entraînées par ces changements. Ce type de modèle offre cinq débouchés principaux pour l’industrie pharmaceutique : (1) l’identification de nouveaux modes de traitement physiologiquement ciblés ; (2) le sauvetage de molécules rencontrant de sérieuses difficultés en phase d’essais cliniques ; (3) la découverte de nouvelles applications pour des molécules existantes ; (4) l’identification des critères clés d’exclusion entraînant une amélioration sensible des taux de succès en phase clinique ; et (5) l’identification de bio-marqueurs permettant le suivi en temps réel des effets d’une intervention.
Références
- Lusis AJ. A thematic review series: systems biology approaches to metabolic and cardiovascular disorders. J Lipid Res 2006; 22 : 1268–74. [Google Scholar]
- Oliver SG. From genomes to systems: the path with yeast. Philos Trans R Soc Lond B Biol Sci 2006; 361 : 477–82. [Google Scholar]
- Huan T, Sivachenko AY, Harrison SH, Chen JY. ProteoLens: a visual analytic tool for multi-scale database-driven biological network data mining. BMC Bioinformatics 2008; 9 : S5. [Google Scholar]
- Soppa J. From genomes to function: haloarchaea as model organisms. Microbiology 2006; 152 : 585–90. [Google Scholar]
- Jamshidi N, Palsson BO. Formulating genome-scale kinetic models in the post-genome era. Mol Syst Biol 2008; 4 : 1268–74. [Google Scholar]
- Mete M, Tang F, Xu X, Yuruk N. A structural approach for finding functional modules from large biological networks. BMC Bioinformatics 2008; 9 : S19. [Google Scholar]
- Gadal F, Bozic C, Pillot-Brochet C, et al. Integrated transcriptome analysis of the cellular mechanisms associated with Ha-ras-dependent malignant transformation of the human breast epithelial MCF7 cell line. NAR 2003; 19 : 5789–804. [Google Scholar]
- Gadal F, Starzec A, Bozic C, et al. Integrative analysis of gene expression patterns predicts specific modulations of defined cell functions by estrogen and tamoxifen in MCF7 breast cancer cells. J Mol Endocrinol 2005; 34 : 61–75. [Google Scholar]
- Iris F, Xavier F. De la modélisation cellulaire aux systèmes intégrés. Ecrin 2003; 53 : 13–6. [Google Scholar]
- Iris F. Les modèles physiologiques intégrés. In : Écrin, eds. Technologies du futur, enjeux de société. Paris : Omniscience, 2005 : 40–65. [Google Scholar]
- Iris F, Géa M, Lampe PH, et al. Integrative biology in the discovery of relevant biomarkers monitoring cognitive disorders pathogenesis and progression. BioTribune 2008; 28 : 8–23. [Google Scholar]
- Iris F. Biological modeling in the discovery and validation of cognitive dysfunctions biomarkers. In : Turck CW, ed. Biomarkers for psychiatric disorders. Berlin : Springer Verlag, 2008 : 486–538. [Google Scholar]
- Salhi I, Cambon-Roques S, Lamarre I, et al. The anti-Müllerian hormone type II receptor: insights into the binding domains recognized by a monoclonal antibody and the natural ligand. Biochem J 2004; 379 : 785–93. [Google Scholar]
- Xavier F, Allard S. Anti-Müllerian hormone, beta-catenin and Müllerian duct regression. Mol Cell Endocrinol 2003; 211 : 115–21. [Google Scholar]
- Wolff E, Lutz-Ostertag Y. Histological phenomenon of the regression of the Müllerian ducts in the male chick embryo. Arch Anat Histol Embryol 1952; 34 : 459–65. [Google Scholar]
- Orvis GD, Jamin SP, Kwan KM, et al. Functional redundancy of TGF-beta family type I receptors and receptor-Smads in mediating anti-Mullerian hormone-induced Mullerian duct regression in the mouse. Biol Reprod 2008; 78 : 994–1001. [Google Scholar]
- Klattig J, Englert C. The Müllerian duct: recent insights into its development and regression. Sex Dev 2007; 1 : 271–8. [Google Scholar]
Liste des figures
|
Figure 1. Approche événementielle et relativiste de l’information. A. Le processus d’intégration commence par la définition du problème biomédical devant être traité (cancer du sein dépendant de l’activation de l’oncogène ras, hypercholestérolémie familiale, détoxification d’une molécule déterminée, etc.), ce qui permet de définir des contextes physiopathologiques. B. Toutes les informations stockées dans les bases de données publiques ayant un lien quelconque avec ces contextes sont rapatriées et structurées sous forme de base de données indexée (en termes de composantes biologiques, d’interactions, d’interdépendances, etc.). Si des données expérimentales sont disponibles, elles sont injectées dans cette base de données et sont prises en compte dans une nouvelle indexation. C. Les arborescences de l’index sont ensuite visualisées et les éléments donnant naissance aux embranchements sont utilisés pour générer des hypothèses fonctionnelles. D. Chaque hypothèse ainsi générée est alors testée contre l’ensemble des bases de données publiques, le but recherché ici étant la destruction de l’hypothèse. E. Les éléments d’information permettant de la détruire sont alors utilisés, conjointement avec les éléments d’arborescence, pour générer une nouvelle hypothèse qui est à son tour soumise au processus de destruction et ainsi de suite, de façon itérative, jusqu’à obtention d’une hypothèse ne pouvant pas être détruite. F. Les hypothèses non détruites accumulées sont alors intégrées en méta-hypothèses qui sont à leur tour soumises au même processus de destruction. Ce procédé, qui est étonnamment rapide (moins d’un an pour une maladie complexe et largement incomprise telle que la maladie de Creutzfeldt-Jakob), amène à la construction d’un modèle biologique, souvent très détaillé et directement vérifiable par expérimentations indépendantes. |
| Dans le texte | |
|
Figure 2. Observations expérimentales d’origine associées au processus de régression du canal de Müller chez la souris. Ces données ont été obtenues par le Pr F. Xavier (CNRS et ECP) et son équipe. Deux voies principales sont activées dans les cellules mésenchymateuses en réponse à l’AMH. La voie PI3K, qui aboutit à une accumulation cytoplasmique massive de β-caténine, et la voie Smad, qui résulte de la présence de complexes composés des protéines Smad4, β-caténine et Lef dans le noyau de ces cellules. Les points d’interrogation signifient l’incertitude quant à la forme structurale du récepteur à l’AMH entraînant l’activation de ces voies de signalisation (le récepteur AMHRII seul ou sa forme complexe AMHRII-ALK). |
| Dans le texte | |
|
Figure 3. Résultats simplifiés de modélisation décrivant les conditions requises pour l’expression d’AMHRII dans les cellules mésenchymateuses. Ce diagramme décrit les communications entre cellules épithéliales (peu nombreuses) et cellules mésenchymateuses ainsi que l’état physiologique de ces cellules au moment où la réponse à l’AMH va commencer. Les lignes rouges indiquent les mécanismes et les interconnexions qui se trouvent interdites dans ce contexte. |
| Dans le texte | |
|
Figure 4A Résultats simplifiés de modélisation décrivant la première phase de réponse à l’AMH dans les cellules mésenchymateuses. Ici, seul le récepteur AMHRII est actif (imposé par des effets de fluidique membranaire). Le signal résultant est l’activation de certains éléments de la voie PI3K, aboutissant à l’accumulation d’une fraction de la β-caténine cellulaire et la sécrétion de fibronectine, préparant la rupture des contacts entre cellules mésenchymateuses. Ces événements concernent principalement les cellules à la partie superficielle des contacts avec le système vasculaire dans le manchon (voir texte), les cellules des couches internes n’ayant pas encore un accès libre à l’hormone. Les lignes et croix rouges indiquent des voies devenues interdites. Les points d’interrogation indiquent l’incertitude quant à l’identité exacte de la composante correspondante. Les boîtes bleues correspondent aux éléments vérifiés in vivo chez la souris par F. Xavier et son équipe au CNRS. |
| Dans le texte | |
|
Figure 4B Résultats simplifiés de modélisation décrivant la seconde phase de réponse à l’AMH dans les cellules mésenchymateuses. Les complexes AMHRII-ALK sont maintenant activés alors que AMHRII seul reste encore fonctionnel. Il en résulte la mobilisation et la stabilisation de la β-caténine membranaire, entraînant la décomposition des contacts entre cellules mésenchymateuses, et la ré-induction partielle des voies Smad qui étaient jusqu’alors inhibées. À ce stade, les cellules mésenchymateuses superficielles se dissocient et changent de forme (flèches annotées « 2 »). Les cellules des couches internes du manchon sont maintenant exposées à l’AMH et entrent en phase 1 (flèches annotées « 1 »). Les boîtes entourées d’un cadre gras correspondent aux éléments vérifiés in vivo chez la souris par F. Xavier et son équipe au CNRS. |
| Dans le texte | |
|
Figure 4C Résultats simplifiés de modélisation décrivant la troisième phase de réponse à l’AMH dans les cellules mésenchymateuses. En réponse à la décomposition des contacts entre cellules (phase 2), toutes les cellules mésenchymateuses, sur la profondeur du manchon, sont localement exposées à l’AMH et les complexes AMHRII-ALK sont maintenant les récepteurs majoritairement actifs. Cela entraîne la réactivation complète des voies Smad, la sécrétion d’enzymes (MMP3 et 9) et la dégradation partielle de la membrane fibreuse séparant les cellules épithéliales des cellules mésenchymateuses (flèches annotées 3 et 4). Ces dernières sont protégées des effets de ces enzymes par la fibronectine sécrétée durant les phases 1 et 2. En retour, la dégradation partielle de la membrane fibreuse induit (1) la mort des cellules épithéliales ancrées à des fibres dégradées et (2) la conversion en cellules mésenchymateuses de celles qui sont ancrées à des régions de la membrane encore intactes. Les croix rouges indiquent des interactions devenues interdites. L’orientation des flèches indique des gènes dont l’expression est fortement augmentée (pointe vers le haut) et fortement inhibée (pointe vers le bas). Les boîtes entourées d’un cadre gras correspondent aux éléments vérifiés in vivo chez la souris par le F. Xavier et son équipe au CNRS. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.