")
")
| Issue |
Med Sci (Paris)
Volume 25, Number 5, Mai 2009
Évaluation des risques et perspectives thérapeutiques en oncologie colorectale
|
|
|---|---|---|
| Page(s) | 489 - 496 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/2009255489 | |
| Published online | 15 mai 2009 | |
Le facteur de croissance des fibroblastes 23 et son récepteur Klotho
Un nouvel axe de régulation du bilan du phosphate
Fibroblast Growth Factor 23-Klotho: a new axis of phophate balance control
1
Service d’explorations fonctionnelles, Bâtiment Sèvres 8e étage, AP-HP, Hôpital Necker-Enfants alades, 149, rue de Sèvres, 75015 Paris, France.
2
Centre de Recherche Croissance et Signalisation (Inserm U845), Université Paris Descartes, Faculté de Médecine, Paris, France
3
Service de Néphrologie et Dialyse. Clinique du Landy, Saint-Ouen, France
4
Service de Physiologie et Radio-isotopes, Hôpital Européen Georges Pompidou, AP-HP, Paris, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Le rein tient une place centrale dans la détermination de la phosphatémie. Il adapte la réabsorption de phosphate dans le tubule proximal aux besoins de l’organisme et il contrôle l’absorption digestive de phosphate et de calcium par l’intermédiaire du calcitriol qu’il synthétise. La découverte du FGF23 (fibroblast growth factor 23) et son identification comme une hormone qui contrôle le métabolisme du phosphate et du calcitriol a permis de préciser les mécanismes par lesquels les fonctions de réabsorption du phosphate et de synthèse hormonale dans le tubule proximal rénal sont couplées. L’identification du FGF23 a mis en évidence un axe os-rein qui contrôle la minéralisation osseuse. L’étude de modèles animaux a considérablement amélioré notre compréhension de l’homéostasie du phosphate et a fait émerger le rôle de la protéine klotho qui est indispensable à l’action du FGF23. Cette revue détaille les fonctions du FGF23 et de klotho en physiologie et au cours de différentes maladies d’origine génétique ou acquise. Le phosphate est impliqué dans les processus de calcification vasculaire et tissulaire, et de prolifération cellulaire. Les anomalies touchant l’axe FGF23-Klotho altèrent l’espérance de vie et sont impliquées dans les processus de vieillissement.
Abstract
The kidney is a key player of phosphate balance, it determines serum phosphate levels by coupling phosphate reabsorption in the renal proximal tubule, calcitriol synthesis and consequently intestinal phosphate absorption. The identification of fibroblast growth factor 23 (FGF23) as a hormone regulating phosphate and calcitriol metabolism has unveiled the mechanisms that coordinate these renal proximal tubule functions. A bone-kidney axis has emerged that controls bone mineralization. Animal model studies have improved our understanding of phosphate homeostasis and revealed the role of the Klotho protein, which is mandatory to FGF23 action. In this review, we detail FGF23 and Klotho implication in physiology and in genetic or acquired disorders. Phosphate ion is involved in vascular and soft tissue calcification and is important for cell proliferation. Disorders of FGF23-klotho axis alter life span and are involved in senescence.
© 2009 médecine/sciences - Inserm / SRMS
Le phosphate est un anion indispensable au fonctionnement cellulaire et à une minéralisation osseuse correcte. Un excès de phosphate circulant est responsable de l’apparition de calcifications tissulaires et vasculaires qui diminuent l’espérance de vie. Un déficit en phosphate diminue la minéralisation osseuse et perturbe le fonctionnement cellulaire [1]. Le rein joue un rôle central dans l’homéostasie du phosphate, il adapte en permanence les sorties aux entrées de façon à maintenir constantes dans un organisme adulte la phosphatémie et la quantité de phosphate. L’hormone parathyroidienne (PTH) peut diminuer la phosphatémie en augmentant l’élimination urinaire de phosphate. Cependant, le paramètre qui règle la sécrétion de cette hormone est la calcémie ionisée, et non pas directement la phosphatémie. L’existence de facteurs stimulant l’élimination urinaire de phosphate autres que la PTH a été postulée il y a plusieurs décennies devant des tableaux d’hypophosphatémie profonde liée à une excrétion urinaire de phosphate inappropriée et que n’expliquait pas une hyperparathyroïdie. Ces maladies peuvent être acquises, par exemple les tumeurs ostéomalaciantes, ou être d’origine génétique comme le rachitisme hypophosphatémique autosomique dominant. L’étude de ces deux maladies a permis d’identifier le premier facteur phosphaturiant : le FGF 23 (fibroblast growth factor 23). D’autres facteurs moins bien caractérisés qui augmentent également l’élimination urinaire de phosphate : le FGF7, sFRP4 (secreted frizzled-related protein 4) ne seront pas présentés dans cette revue.
Caractéristiques biochimiques du FGF23
Le FGF231 est un peptide de 251 acides aminés qui est présent dans le plasma des sujets sains. Son ARN messager est exprimé dans le cœur, le foie, le bloc thyro-parathyroïdien et surtout l’os dans les ostéocytes et les ostéoblastes [2, 3]. Dans le plasma, le FGF23 est détecté sous 2 formes : un peptide de 32kD qui correspond à la forme intacte et dont la concentration plasmatique est du même ordre que celle de la PTH (inférieure à 50 pg/ml avec les techniques de dosage actuelles) et deux peptides résultant du clivage enzymatique de la forme intacte au niveau d’un site particulier situé entre les acides aminés 176 et 178. Ce clivage libère un peptide amino-terminal et un peptide carboxy-terminal. Il semble que seule la forme intacte du peptide ait une activité biologique. Le lieu de clivage du FG23 ainsi que l’enzyme responsable n’ont pas encore été identifiés mais il est bien établi qu’une glycosylation anormale du FGF23 altère sa stabilité [4].
La demi-vie plasmatique du FGF23 est courte (quelques heures) comme le suggèrent la diminution et la normalisation rapides de sa concentration après l’exérèse d’une tumeur sécrétante [5, 6].
Contrôle de la sécrétion de FGF23
Trois paramètres modifient la concentration plasmatique de FGF23 : la phosphatémie, les apports digestifs en phosphate et la calcitriolémie. Une augmentation des apports digestifs en phosphate accroît, chez l’homme, la concentration plasmatique de FGF23. À l’opposé, une restriction de ces apports s’accompagne d’une diminution de la concentration de FGF23 [7, 8].
Le calcitriol est un puissant régulateur de la production et de la concentration plasmatique de FGF23. L’injection de calcitriol chez des patients dialysés [9] et chez la souris augmente la concentration plasmatique de FGF23 et l’expression de son ARN messager dans l’os [10, 11]. Cet effet du calcitriol est vraisemblablement lié à la présence de séquences de réponse à la vitamine D (VDRE) dans le promoteur du FGF23.
Lorsque le débit de filtration glomérulaire diminue, l’on observe de façon précoce une augmentation de la concentration plasmatique de FGF23 qui devient très élevée dans l’insuffisance rénale terminale [12–14]. Cette augmentation permettrait de s’opposer à l’augmentation de la phosphatémie lors de la baisse du débit de filtration glomérulaire. Il est également possible que l’augmentation du FGF23, qui touche aussi bien la forme intacte que les peptides de dégradation, soit en partie secondaire à une diminution du catabolisme de la protéine. Cependant, la diminution de la phosphatémie liée au traitement au cours de l’insuffisance rénale chronique s’accompagne d’une baisse de la concentration plasmatique du FGF23.
Effets biologiques du FGF23
La surexpression de FGF23
La surexpression de FGF23 chez l’homme et dans différents modèles animaux entraîne de façon constante une hypophosphatémie - due à une augmentation des pertes urinaires en phosphate témoignant d’un défaut de réabsorption tubulaire touchant spécifiquement le phosphate - ainsi qu’une ostéomalacie ou un rachitisme, des douleurs osseuses et musculaires, et une asthénie. La fuite urinaire de phosphate s’explique par la diminution de l’expression de l’ARN messager des cotransporteurs rénaux sodium/phosphate NPT2a et NPT2c qui se traduit par une diminution de l’abondance des protéines correspondantes dans la bordure en brosse des cellules tubulaires proximales [15].
La surexpression de FGF23 s’accompagne également d’une baisse rapide de la calcitriolémie qui s’explique par la diminution de l’expression et de l’activité enzymatique de la 1 alpha hydroxylase rénale2. Le FGF23 stimule également l’expression et l’activité de la 24 hydroxylase, l’enzyme qui dégrade le calcitriol en 1, 24-25 hydroxyvitamine D [16, 17].
La diminution de l’absorption digestive de phosphate observée après injection de FGF23 chez les animaux s’explique exclusivement par la baisse de la calcitriolémie, et non par un effet direct du FGF23 sur le transporteur intestinal de phosphate NPT2b. Cet effet n’existe pas chez les animaux invalidés pour le récepteur de la vitamine D [17].
L’injection de FGF23 chez l’animal induit une diminution rapide de l’expression de l’ARN messager de la PTH dans les glandes parathyroïdiennes et une baisse de la concentration de PTH circulante [18]. Contrairement à ce qui est observé dans le rein, le FGF23 stimule l’expression de la 1-alpha-hydroxylase dans les glandes parathyroïdes [19]. L’augmentation locale de la synthèse de calcitriol qui en résulte renforcerait l’effet inhibiteur du FGF23 sur la production de PTH.
Maladies associées à une production excessive de FGF23
La production non contrôlée de FGF23 se rencontre au cours de plusieurs maladies. Les tumeurs ostéomalaciantes sont généralement dérivées de tumeurs mésenchymateuses, le plus souvent bénignes et parfois de petite taille, ce qui rend leur détection difficile et le recours à différentes techniques de scintigraphie parfois nécessaire.
Dans le rachitisme autosomique dominant, une mutation du site de clivage du FGF23 le rend résistant à la dégradation enzymatique [3, 20].
Le rachitisme hypophosphatémique lié au chromosome X est dû à une mutation dans un gène codant pour une protéine dont la structure la rapproche des endopeptidases (PHEX, phosphate regulating endopeptidase homolog, X-linked). On a cru initialement que cette enzyme potentielle PHEX était capable de dégrader le FGF23 et donc que les mutations de PHEX favorisaient l’accumulation de FGF23. PHEX est effectivement exprimée avec FGF23 dans les ostéocytes et la concentration plasmatique de FGF23 est augmentée lorsqu’existe une mutation de PHEX. Cependant il n’est pas confirmé que le FGF23 soit un substrat de PHEX, et les mécanismes de la surproduction osseuse de FGF23 dans cette pathologie restent obscurs ou spéculatifs.
La dentin matrix protein 1 (DMP1) est à la fois un facteur transcriptionnel et un peptide sécrété qui joue un rôle important dans la différenciation des ostéoblastes. Des mutations de ce gène sont responsables d’une hypophosphatémie autosomique récessive et s’accompagnent d’une augmentation du FGF23 par un mécanisme inconnu.
Dans environ 50 % des fibrodysplasies osseuses de Mc Cune Albright3 on observe une hypophosphatémie liée à une surproduction de FGF23 à partir des cellules dysplasiques. Cette maladie est due à une mutation post-zygotique d’une petite protéine G (Gs α) aboutissant à des anomalies de prolifération des cellules osseuses dont certaines synthétisent du FGF23 de façon non régulée.
Diminution de la production de FGF23
La baisse de concentration du FGF23, chez l’homme comme chez la souris - dans cette dernière en réponse à l’invalidation du gène codant pour le FGF23 - entraîne une hyperphosphatémie, une hypercalcémie, une hypercalcitriolémie, une baisse de la concentration de la PTH plasmatique, l’apparition de dépôts phosphocalciques dans les tissus mous, et chez la souris, un vieillissement accéléré (baisse de la masse musculaire, perte des poils, hypogonadisme, emphysème pulmonaire, atrophie cutanée) [21, 22].
Une concentration basse inappropriée de FGF23 s’observe chez l’homme au cours de mutations touchant les sites de glycosylation du FGF23 ou l’enzyme GALNT3 (mucin-like glycosyltransferase GalNAc transferase-3) qui glycosyle le FGF23. Une glycosylation anormale du FGF23 augmente sa dégradation et diminue donc la concentration de FGF23 intact circulant alors que celle du peptide carboxy-terminal est augmentée [4]. Ces observations confirment que l’activité biologique du FGF23 est essentiellement due au peptide intact.
Dans la calcinose tumorale, le contrôle de la phosphatémie par des chélateurs digestifs du phosphate et l’induction d’une phosphaturie par des inhibiteurs de l’anhydrase carbonique permettent de faire régresser les calcifications tumorales. Cet effet de la baisse de la phosphatémie sur les calcifications suggère le rôle prépondérant de l’hyperphosphatémie sur la formation des calcifications, ce que confirment des modèles animaux. La double invalidation des gènes codant le FGF23 et la 1 alpha hydroxylase normalise le phénotype des souris invalidées pour le FGF23, mais corrige à la fois l’hypercalcitriolémie et l’hyperphosphatémie [21], ce qui ne permet pas de trancher entre les rôles respectifs de ces deux anomalies dans la genèse des calcifications. La correction spécifique de l’hypercalcitriolémie sans modification de l’hyperphosphatémie, obtenue en soumettant des souris Fgf23−/− à un régime pauvre en vitamine D, permet un allongement de la survie des animaux mais les calcifications vasculaires persistent. À l’opposé, la normalisation de la phosphatémie avec persistance de l’hypercalcitriolémie obtenue en soumettant les souris Fgf23−/− à un régime pauvre en phosphate s’accompagne d’une normalisation complète du phénotype, suggérant le rôle délétère prépondérant de l’hyperphosphatémie dans les anomalies observées [23].
Le récepteur du FGF23, la protéine Klotho
Le FGF23 se lie avec une affinité modeste aux différents récepteurs des FGF (FGFR) suggérant qu’un cofacteur pourrait être nécessaire à la liaison du FGF23 à un FGFR. L’insertion non intentionnelle chez des souris d’un transgène dans le gène codant pour la protéine Klotho, puis l’invalidation ciblée du gène ont permis de mettre en évidence le rôle de cette protéine dans la signalisation du FGF23. Les souris déficientes en Klotho ont un phénotype identique à celui des souris Fgf23−/−, mais avec une concentration de FGF23 plasmatique élevée [24, 25]. Klotho est une protéine de 1014 acides aminés avec un long domaine amino-terminal extracellulaire et un domaine transmembranaire et carboxy-terminal intracellulaires très courts. Le domaine extracellulaire est composé de deux régions homologues KL1 et KL2. Klotho est exprimée à la surface des cellules, mais est également présente dans le plasma sous deux formes : l’une contient les domaines KL1 et KL2 et provient probablement du clivage de la forme membranaire, et l’autre, une protéine de 549 acides aminés, ne contient que le domaine KL1 et provient d’un épissage différentiel de l’exon 3 aboutissant à un peptide directement sécrété (Figure 2A). La forme transmembranaire de Klotho se lie au FGF23 [26, 27] (Figure 2B). L’injection d’anticorps anti-Klotho chez la souris abolit cette interaction et reproduit le phénotype de souris Klotho−/− et Fgf23−/− [27]. La liaison de Klotho au FGF23 augmente l’affinité de ce dernier pour les FGFR ce qui active la phosphorylation du complexe ERK1/2.
 |
Figure 1. FGF23 et l’axe rein-os. Le FGF23 est synthétisé par l’os en réponse à l’augmentation de la phosphatémie. Le FGF23 contrôle l’activité des transporteurs rénaux de phosphate et la synthèse de calcitriol par le tubule proximal et l’absorption intestinale de phosphate par l’intermédiaire de la calcitriolémie. Le FGF23 modifie également la synthèse et la sécrétion de PTH par les glandes parathyroïdiennes. |
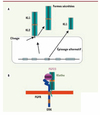 |
Figure 2. A. Les différentes formes de Klotho (KL), la forme transmembranaire, et les deux formes circulantes solubles dont l’une résulte d’un épissage alternatif au niveau du 3e exon du gène. B. Interaction entre Klotho, le récepteur du FGF, et le FGF23 à la surface cellulaire. |
Klotho est exprimée dans un nombre restreint d’organes dont le rein et la glande parathyroïde. De façon étonnante, dans le rein, Klotho est exprimée dans le tubule distal et non dans le tubule proximal, siège du transport de phosphate et de la synthèse de calcitriol. Actuellement, l’on ignore par quel mécanisme le FGF23 modifie l’expression des transporteurs de phosphate, de la 1-alpha-hydroxylase et de la 24-hydroxylase, et la réabsorption de phosphate dans le tubule proximal. Le tubule distal exprime les récepteurs FGFR1 et FGFR3. Klotho aurait une affinité plus importante pour le FGFR1(IIIc) que pour les autres FGFR [27], mais l’invalidation du FGFR1, létale in utero chez la souris, ne permet pas de confirmer son rôle dans la fonction du FGF23. Les mutations activatrices du FGFR1 chez l’homme s’accompagnent d’une hypophosphatémie, d’une concentration circulante inappropriée de calcitriol, mais également d’une concentration plasmatique de FGF23 augmentée [28]. L’invalidation du FGFR3 ne s’accompagne pas d’anomalies du bilan du phosphate [29]. Chez la souris hyp, qui présente une baisse d’expression de PHEX et une augmentation de la concentration plasmatique de FGF23, l’invalidation du FGFR3 ne normalise pas la phosphatémie. Cependant, l’on observe chez ces souris une aggravation de l’hypersécrétion de FGF23 [29].
Dans le tubule rénal distal, l’expression de Klotho stabiliserait l’expression membranaire du canal calcique TRPV5 (transient receptor potential vanilloid 5) grâce à sa faible activité β-glucuronidase [30]. L’importance physiologique de cette propriété qui est indépendante du FGF23 n’est pas établie.
La fonction de la forme soluble circulante de Klotho est inconnue. Cette protéine possède les domaines KL1 et KL2, a une faible activité glucuronidase et est capable de lier le FGF23. Cependant, en l’état actuel de nos connaissances, il est difficile de concevoir que la liaison de Klotho circulante et du FGF23 permette l’interaction avec des FGFR car dans cette hypothèse, toutes les cellules qui expriment un FGFR deviendraient potentiellement sensibles au FGF23.
Les conséquences de la surexpression de Klotho sur le bilan phosphocalcique chez la souris n’ont pas encore été publiées. En revanche, la surexpression de Klotho augmente significativement la durée de vie chez la souris, réprime la voie de signalisation de l’IGF (insulin growth factor) et de l’insuline et réduit l’expression de marqueurs du stress oxydatif [31, 32].
Implication du FGF23 et de Klotho dans des pathologies humaines acquises
Lorsque le débit de filtration glomérulaire (DFG) décroît, la concentration plasmatique de FGF23 intact augmente [12, 13] et l’expression de la forme membranaire de Klotho dans le rein et de la forme circulante KL1 diminue [33]. L’augmentation du FGF23 peut refléter une baisse du catabolisme rénal du peptide ou une adaptation physiologique à la baisse d’expression de Klotho ou à l’accumulation de phosphate. Cette dernière possibilité est confortée par la corrélation qui existe chez le patient insuffisant rénal chronique entre la concentration plasmatique de FGF23 et la phosphatémie, l’excrétion urinaire de phosphate et la corrélation inverse avec les concentrations circulantes de PTH et de calcitriol [12, 14]. L’augmentation précoce du FGF23 lorsque le DFG baisse préviendrait l’hyperphosphatémie en diminuant la réabsorption rénale de phosphate et l’absorption digestive de phosphate grâce à la baisse de la calcitriolémie. Cela a également pour conséquence de diminuer l’absorption digestive de calcium et d’entraîner ainsi une hyperparathyroïdie secondaire.
Au cours de la maladie rénale chronique, avant l’entrée en dialyse, la concentration plasmatique de FGF23 semble prédire la rapidité de dégradation de la fonction rénale chez des sujets non diabétiques [34]. Chez les patients dialysés, la concentration plasmatique de FGF23 est très élevée et prédit la survenue d’une hyperparathyroïdie réfractaire [35, 36] et elle est associée à une mortalité accrue durant la première année de dialyse indépendamment de la valeur de la phosphatémie [37].
Maladies génétiques liées à une réabsorption rénale inappropriée de phosphate et à une anomalie de la phosphatémie chez l’homme. Intacte : peptide non clivé (32kD) ; C-terminal: peptide carboxy-terminal résultant du clivage de la forme intacte ; GALNT3, UDP-N-acetyl-alpha-D-galactosamine:polypeptide N-acetylgalactosaminyltransferase 3 ; DMP1 ; PHEX, phosphate regulating endopeptidase homolog, X-linked ; GNAS, α-subunit of stimulatory G proteins ; NHERF1, Na/H exchange regulatory factor-1 ; NPT2c, type IIc sodium-dependent phosphate transporter.
La possibilité d’un effet direct du FGF23 sur l’os est encore controversée. Dans une étude menée chez les patients dialysés, nous n’avons pas trouvé de corrélation entre la concentration de FGF23 et la densité minérale osseuse ce qui suggère l’absence d’effet direct du FGF23 sur l’os chez ces patients [14].
Après transplantation rénale, il n’est pas rare d’observer une hypophosphatémie en l’absence d’hyperparathyroïdie. Ces anomalies pourraient être liées à une hypersécrétion de FGF23 persistant de façon autonome au-delà de la dialyse [38], et définissant un tableau d’hyperphosphatémie autonomisé.
Il n’existe pas de données chez l’homme suggérant que la surexpression de Klotho puisse protéger contre la dégradation de la fonction rénale, mais dans un modèle murin de glomérulonéphrite, Klotho diminue la sévérité des lésions rénales et l’altération du débit de filtration glomérulaire [39]. La surmortalité observée chez les sujets dialysés ayant des concentrations élevées de FGF23 n’a pas actuellement été reliée à un déficit d’expression de Klotho.
Des polymorphismes du gène codant pour Klotho ont été associés chez l’homme à la longévité, aux risques de survenue de calcifications vasculaires et à la valeur de la densité minérale osseuse après la ménopause [4].
Conclusions
Nos connaissances sur les mécanismes qui permettent d’éviter de grandes variations de la phosphatémie se sont considérablement accrues ces dernières années. L’identification d’une nouvelle hormone, le FGF23, et de son corécepteur Klotho a permis de mieux comprendre la physiopathologie de différentes maladies avec hyper- ou hypophosphatémie (Tableau I), ainsi que la genèse de l’hyperparathyroïdie secondaire au cours de l’insuffisance rénale. De nouveaux traitements interférant avec l’axe FGF23-Klotho devraient être développés dans les prochaines années et permettre un meilleur contrôle de la phosphatémie chez les patients, évitant ainsi la surmorbidité et peut être la surmortalité associées à ces désordres.
La grande analogie de séquence de ce peptide avec celle des autres FGF lui a valu son nom, mais qui ne renseigne pas sur sa fonction.
Cette enzyme est nécessaire à la conversion de la 25-OH vitamine D en calcitriol.
Le syndrome de McCune-Albright (SMA) est classiquement défini par une triade clinique comportant une dysplasie fibreuse des os (DFO), des taches cutanées café au lait et une puberté précoce (PP). La prévalence est estimée entre 1/100 000 et 1/1 000 000. La DFO peut impliquer un ou plusieurs sites squelettiques et elle se manifeste par une boiterie et/ou une douleur et, parfois, par une fracture pathologique. On trouve aussi d’autres endocrinopathies, et une atteinte rénale est observée chez environ 50 % des patients (site orphanet).
Références
- Prie D, Beck L, Urena P, Friedlander G. Recent findings in phosphate homeostasis. Curr Opin Nephrol Hypertens 2005; 14 : 318–24. [Google Scholar]
- Mirams M, Robinson BG, Mason RS, Nelson AE. Bone as a source of FGF23: regulation by phosphate ? Bone 2004; 35 : 1192–9. [Google Scholar]
- ADHR c. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet 2000; 26 : 345–8. [Google Scholar]
- Prie D, Urena P, Friedlander G. Latest findings in phosphate homeostasis. Kidney Int 2009; 4 février online. [Google Scholar]
- Ward LM, Rauch F, White KE, et al. Resolution of severe, adolescent-onset hypophosphatemic rickets following resection of an FGF-23-producing tumour of the distal ulna. Bone 2004; 34 : 905–11. [Google Scholar]
- Yamazaki Y, Okazaki R, Shibata M, et al. Increased circulatory level of biologically active full-length FGF-23 in patients with hypophosphatemic rickets/osteomalacia. J Clin Endocrinol Metab 2002; 87 : 4957–60. [Google Scholar]
- Burnett SM, Gunawardene SC, Bringhurst FR, et al. Regulation of C-terminal and intact FGF-23 by dietary phosphate in men and women. J Bone Miner Res 2006; 21 : 1187–96. [Google Scholar]
- Ferrari SL, Bonjour JP, Rizzoli R. Fibroblast growth factor-23 relationship to dietary phosphate and renal phosphate handling in healthy young men. J Clin Endocrinol Metab 2005; 90 : 1519–24. [Google Scholar]
- Nishi H, Nii-Kono T, Nakanishi S, et al. Intravenous calcitriol therapy increases serum concentrations of fibroblast growth factor-23 in dialysis patients with secondary hyperparathyroidism. Nephron Clin Pract 2005; 101 : c94–9. [Google Scholar]
- Liu S, Tang W, Zhou J, et al. Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin D. J Am Soc Nephrol 2006; 17 : 1305–15. [Google Scholar]
- Kolek OI, Hines ER, Jones MD, et al. 1alpha,25-Dihydroxyvitamin D3 upregulates FGF23 gene expression in bone: the final link in a renal-gastrointestinal-skeletal axis that controls phosphate transport. Am J Physiol Gastrointest Liver Physiol 2005; 289 : G1036–42. [Google Scholar]
- Gutierrez O, Isakova T, Rhee E, et al. Fibroblast growth factor-23 mitigates hyperphosphatemia but accentuates calcitriol deficiency in chronic kidney disease. J Am Soc Nephrol 2005; 16 : 2205–15. [Google Scholar]
- Larsson T, Nisbeth U, Ljunggren O, Juppner H, Jonsson KB. Circulating concentration of FGF-23 increases as renal function declines in patients with chronic kidney disease, but does not change in response to variation in phosphate intake in healthy volunteers. Kidney Int 2003; 64 : 2272–9. [Google Scholar]
- Urena Torres P, Friedlander G, de Vernejoul MC, Silve C, Prie D. Bone mass does not correlate with the serum fibroblast growth factor 23 in hemodialysis patients. Kidney Int 2008; 73 : 102–7. [Google Scholar]
- Shimada T, Urakawa I, Yamazaki Y, et al. FGF-23 transgenic mice demonstrate hypophosphatemic rickets with reduced expression of sodium phosphate cotransporter type IIa. Biochem Biophys Res Commun 2004; 314 : 409–14. [Google Scholar]
- Shimada T, Mizutani S, Muto T, et al. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci USA 2001; 98 : 6500–5. [Google Scholar]
- Saito H, Kusano K, Kinosaki M, et al. Human fibroblast growth factor-23 mutants suppress Na+-dependent phosphate co-transport activity and 1alpha,25-dihydroxyvitamin D3 production. J Biol Chem 2003; 278 : 2206–11. [Google Scholar]
- Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V, et al. The parathyroid is a target organ for FGF23 in rats. J Clin Invest 2007; 117 : 4003–8. [Google Scholar]
- Krajisnik T, Bjorklund P, Marsell R, et al. Fibroblast growth factor-23 regulates parathyroid hormone and 1alpha-hydroxylase expression in cultured bovine parathyroid cells. J Endocrinol 2007; 195 : 125–31. [Google Scholar]
- Shimada T, Muto T, Urakawa I, et al. Mutant FGF-23 responsible for autosomal dominant hypophosphatemic rickets is resistant to proteolytic cleavage and causes hypophosphatemia in vivo. Endocrinology 2002; 143 : 3179–82. [Google Scholar]
- Razzaque MS, Sitara D, Taguchi T, St-Arnaud R, Lanske B. Premature aging-like phenotype in fibroblast growth factor 23 null mice is a vitamin D-mediated process. Faseb J 2006; 20 : 720–2. [Google Scholar]
- Shimada T, Kakitani M, Yamazaki Y, et al. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest 2004; 113 : 561–8. [Google Scholar]
- Stubbs JR, Liu S, Tang W, et al. Role of hyperphosphatemia and 1,25-dihydroxyvitamin D in vascular calcification and mortality in fibroblastic growth factor 23 null mice. J Am Soc Nephrol 2007; 18 : 2116–24. [Google Scholar]
- Kuro-o M, Matsumura Y, Aizawa H, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997; 390 : 45–51. [Google Scholar]
- Tsujikawa H, Kurotaki Y, Fujimori T, Fukuda K, Nabeshima Y. Klotho, a gene related to a syndrome resembling human premature aging, functions in a negative regulatory circuit of vitamin D endocrine system. Mol Endocrinol 2003; 17 : 2393–403. [Google Scholar]
- Kurosu H, Ogawa Y, Miyoshi M, et al. Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem 2006; 281 : 6120–3. [Google Scholar]
- Urakawa I, Yamazaki Y, Shimada T, et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 2006; 444 : 770–4. [Google Scholar]
- White KE, Cabral JM, Davis SI, et al. Mutations that cause osteoglophonic dysplasia define novel roles for FGFR1 in bone elongation. Am J Hum Genet 2005; 76 : 361–7. [Google Scholar]
- Liu S, Vierthaler L, Tang W, Zhou J, Quarles LD. FGFR3 and FGFR4 do not mediate renal effects of FGF23. J Am Soc Nephrol 2008; 19 : 2342–50. [Google Scholar]
- Chang Q, Hoefs S, van der Kemp AW, et al. The beta-glucuronidase klotho hydrolyzes and activates the TRPV5 channel. Science 2005; 310 : 490–3. [Google Scholar]
- Kurosu H, Yamamoto M, Clark JD, et al. Suppression of aging in mice by the hormone Klotho. Science 2005; 309 : 1829–33. [Google Scholar]
- Yamamoto M, Clark JD, Pastor JV, et al. Regulation of oxidative stress by the anti-aging hormone klotho. J Biol Chem 2005; 280 : 38029–34. [Google Scholar]
- Koh N, Fujimori T, Nishiguchi S, et al. Severely reduced production of klotho in human chronic renal failure kidney. Biochem Biophys Res Commun 2001; 280 : 1015–20. [Google Scholar]
- Fliser D, Kollerits B, Neyer U, et al. Fibroblast growth factor 23 (FGF23) predicts progression of chronic kidney disease: the Mild to Moderate Kidney Disease (MMKD) Study. J Am Soc Nephrol 2007; 18 : 2600–8. [Google Scholar]
- Kazama JJ, Sato F, Omori K, et al. Pretreatment serum FGF-23 levels predict the efficacy of calcitriol therapy in dialysis patients. Kidney Int 2005; 67 : 1120–5. [Google Scholar]
- Nakanishi S, Kazama JJ, Nii-Kono T, et al. Serum fibroblast growth factor-23 levels predict the future refractory hyperparathyroidism in dialysis patients. Kidney Int 2005; 67 : 1171–8. [Google Scholar]
- Gutierrez OM, Mannstadt M, Isakova T, et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med 2008; 359 : 584–92. [Google Scholar]
- Evenepoel P, Naesens M, Claes K, Kuypers D, Vanrenterghem Y. Tertiary hyperphosphatoninism accentuates hypophosphatemia and suppresses calcitriol levels in renal transplant recipients. Am J Transplant 2007; 7 : 1193–200. [Google Scholar]
- Haruna Y, Kashihara N, Satoh M, et al. Amelioration of progressive renal injury by genetic manipulation of Klotho gene. Proc Natl Acad Sci USA 2007; 104 : 2331–6. [Google Scholar]
Liste des tableaux
Maladies génétiques liées à une réabsorption rénale inappropriée de phosphate et à une anomalie de la phosphatémie chez l’homme. Intacte : peptide non clivé (32kD) ; C-terminal: peptide carboxy-terminal résultant du clivage de la forme intacte ; GALNT3, UDP-N-acetyl-alpha-D-galactosamine:polypeptide N-acetylgalactosaminyltransferase 3 ; DMP1 ; PHEX, phosphate regulating endopeptidase homolog, X-linked ; GNAS, α-subunit of stimulatory G proteins ; NHERF1, Na/H exchange regulatory factor-1 ; NPT2c, type IIc sodium-dependent phosphate transporter.
Liste des figures
|
Figure 1. FGF23 et l’axe rein-os. Le FGF23 est synthétisé par l’os en réponse à l’augmentation de la phosphatémie. Le FGF23 contrôle l’activité des transporteurs rénaux de phosphate et la synthèse de calcitriol par le tubule proximal et l’absorption intestinale de phosphate par l’intermédiaire de la calcitriolémie. Le FGF23 modifie également la synthèse et la sécrétion de PTH par les glandes parathyroïdiennes. |
| Dans le texte | |
|
Figure 2. A. Les différentes formes de Klotho (KL), la forme transmembranaire, et les deux formes circulantes solubles dont l’une résulte d’un épissage alternatif au niveau du 3e exon du gène. B. Interaction entre Klotho, le récepteur du FGF, et le FGF23 à la surface cellulaire. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.