")
")
| Issue |
Med Sci (Paris)
Volume 25, Number 3, Mars 2009
|
|
|---|---|---|
| Page(s) | 288 - 292 | |
| Section | Dossier technique | |
| DOI | https://doi.org/10.1051/medsci/2009253288 | |
| Published online | 15 March 2009 | |
Modélisation in vitro du système nerveux par génie tissulaire
Tissue-engineered models of the nervous system
Laboratoire d’organogenèse expérimentale (LOEX), Centre de recherche FRSQ du CHA de Québec, Hôpital du Saint-Sacrement et Département de chirurgie, Faculté de médecine, Université Laval, Québec (Québec) Canada, LOEX, Hôpital du Saint-Sacrement, 1050, chemin Sainte-Foy, Québec (Québec) G1S 4L8 Canada
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
Le système nerveux est un tissu extraordinairement complexe, exposé à des traumatismes et à des maladies dégénératives très difficiles à traiter. Pour faciliter son étude, des systèmes de culture in vitro ont été mis au point depuis de nombreuses années, soit sous la forme de cultures de neurones et de cellules gliales en monocouche, soit sous la forme de cultures organotypiques de tranches de tissu nerveux. Ces deux types de modèles ont permis de faire grandement avancer les connaissances dans le domaine des neurosciences, mais présentent toutefois des limites pour certaines études, le premier risquant d’être trop simplificateur et le second trop complexe. Le génie tissulaire appliqué aux neurosciences propose une nouvelle approche beaucoup plus polyvalente pour modéliser le système nerveux. Celle-ci consiste à reconstruire un ou plusieurs tissus en trois dimensions, en ayant le choix de chacun des éléments, cellulaire ou moléculaire, qui le ou les composent, et en ajustant le niveau de complexité que l’on souhaite atteindre en fonction des besoins. Ainsi, grâce à la technique de modélisation en trois dimensions de la moelle épinière par génie tissulaire à l’aide d’un double compartiment, il a été possible de reproduire, pour la première fois in vitro, la formation de gaines de myéline autour de l’axone de neurones moteurs. Il s’agit d’une percée qui met en évidence le potentiel prometteur du génie tissulaire dans la modélisation du système nerveux in vitro. De plus, ces modèles performants de culture en trois dimensions combinés à l’utilisation de neurones et de cellules gliales humaines adultes différenciés à partir de précurseurs neuraux tirés de tissus provenant des patients (peau, gras, moelle osseuse) ouvre des perspectives très prometteuses pour l’étude des maladies neurodégénératives.
Abstract
The nervous system is extraordinarily complex and exposed to various trauma and degenerative diseases that remain difficult to treat. To facilitate its study, in vitro models were developed by culturing neurons and glial cells in monolayer cultures, or through organotypic cultures of brain or spinal cord slices. These in vitro models were, and are still very helpful for the advancement of neurosciences. However, they are for some studies, either overly simplified, or too complex. The application of tissue engineering to neurosciences offers a new and highly versatile approach to develop accurate models of the nervous system. These models can be engineered in three-dimensions while choosing for each individual component, cellular and molecular, that will compose it. The level of complexity of the model can be adjusted from the simplest to the more complete as needed. For example, through the use of a threedimensional tissue-engineered model of the spinal cord, it was possible to reproduce the process of myelin sheath formation around motor neuron axons for the first time in vitro. This breakthrough shows the promising potential of tissue engineering in the development of powerful in vitro models of the nervous system. The combination of these models with the use of human adult neurons and glial cells obtained from the differentiation of neural precursor cells isolated from accessible tissues from patients (skin, fat, bone marrow), opens promising perspectives to better understand neurodegenerative diseases.
© 2009 médecine/sciences - Inserm / SRMS
Traumatisme crânien, accident vasculaire cérébral, blessure de la moelle épinière, maladies neurodégénératives qui provoquent la destruction ciblée de certains neurones : toute altération de la structure ou du fonctionnement du système nerveux entraîne de graves conséquences sur l’ensemble de l’organisme.
Parmi les maladies neurodégénératives, nombreuses sont celles dont on ignore encore la cause et même le processus évolutif. C’est le cas, en particulier, des maladies pour lesquelles une origine génétique n’a pas encore pu être clairement décelée : maladie de Parkinson, maladie d’Alzheimer, sclérose latérale amyotrophique.
Des modèles animaux reproduisant en partie ou en totalité les symptômes de ces affections ont été mis au point. Ils sont d’une grande utilité pour mieux en comprendre la physiopathologie. Toutefois, l’étude des répercussions du dysfonctionnement du système nerveux sur un organisme entier demeure difficile et n’apporte pas toujours les réponses claires qu’on souhaiterait obtenir. C’est pourquoi, la modélisation in vitro du système nerveux à partir de systèmes de culture sophistiqués mimant le plus fidèlement possible le tissu d’origine permettrait de distinguer les processus biologiques les uns des autres et de déterminer celui ou ceux qui sont ciblés par la maladie pour mieux comprendre ses causes et son fonctionnement.
Les modèles in vitro
L’étude du système nerveux in vitro présente un handicap de taille, puisque, contrairement à la plupart des autres cellules, les neurones ne se multiplient pas. Cela signifie qu’il faudra en prélever à nouveau à chaque expérience sans pouvoir établir de banques et qu’il sera presque impossible d’en obtenir d’origine humaine.
Cultures en monocouche ou double compartiment
La culture in vitro de neurones de tous types (hippocampiques, moteurs, sympathiques, etc.) est réalisée de manière standard depuis de nombreuses années. Que l’on procède par isolement enzymatique des cellules ou par une technique d’explant, ce type de culture permet de mener des études variées et performantes, par exemple, des analyses immunocytochimiques, des mesures d’électrophysiologie ou des tests de toxicité [1].
La principale qualité de ces modèles est d’isoler un type cellulaire, de façon à pouvoir le tester indépendamment de ses partenaires habituels, dans un système de culture très facile à manipuler et très commode à visualiser en imagerie. Il est possible de réaliser des co-cultures de plusieurs types cellulaires afin de suivre les interactions cellules/cellules, mais cette option est limitée à cause du comportement particulier des cellules prolifératives en monocouche : celles-ci ont tendance, en effet, à envahir le flacon de culture, au détriment des cellules non prolifératives que sont les neurones.
Ce handicap peut être surmonté grâce à un système de culture à double compartiment. Ainsi, l’on cultive les neurones au fond d’une boîte de Pétri et les cellules gliales sur un autre support, qui peut être une lamelle de verre retournée et déposée sur les neurones dont elle sera séparée par des espaceurs ; ce système présente l’avantage de permettre une grande proximité des cellules, tout en évitant un contact direct [2]. Une autre technique plus courante consiste à cultiver les cellules gliales dans une chambre de culture séparée insérée dans la boîte de Pétri, et dont le fond est constitué d’une membrane poreuse qui laisse passer les facteurs de croissance mais pas les cellules [3, 4]. La technique de culture à double compartiment offre l’avantage de la simplicité ; de plus, elle permet de récupérer les cellules indépendamment les unes des autres après la culture pour des analyses de biologie moléculaire faciles.
Cependant, un contact direct entre les cellules gliales et les neurones est souvent nécessaire pour qu’un effet soit reproduit in vitro et c’est le cas, par exemple : du recyclage du glutamate par les astrocytes au niveau synaptique ; de la libération locale d’un facteur de croissance qui sera trop dilué et donc inefficace dans une culture à double compartiment ; ou encore d’un milieu conditionné. De plus, les neurones cultivés en monocouche n’ont pas toujours un comportement comparable à celui qu’ils adoptent dans leur tissu d’origine, c’est-à-dire un environnement tridimensionnel (3D) où ils sont entourés de cellules gliales.
Cultures de tranches de tissu nerveux
C’est pourquoi, à titre de complément aux études faites sur les neurones en monocouche, des modèles de culture de tranches de tissu nerveux ont été mis au point. La technique la plus connue est certainement la technique du tube rouleau, qui consiste à fixer un explant de tissu nerveux sur une lame de verre au moyen d’un caillot de plasma de poulet. La lame de verre est alors mise dans une éprouvette de plastique contenant du milieu de culture et placée sur un tambour à rouleau qui tourne à une vitesse précise et selon un angle approprié. Avec cette technique, des tranches de différentes parties du système nerveux central de rongeurs nouveau-nés maintiennent leur organisation organotypique et permettent la visualisation des neurones. De plus, les cellules sont accessibles pour la micromanipulation et elles survivent plusieurs semaines, offrant alors la possibilité de les traiter avec différentes molécules [5–7].
Les explants peuvent aussi être spontanément fixés à une membrane poreuse et transparente afin d’être maintenus à l’interface air-liquide. Le principal avantage de cette technique est de pouvoir réaliser des expériences morphologiques et électrophysiologiques quelques heures seulement après l’explantation, ce qui facilite grandement les études portant sur la réorganisation synaptique et les études consacrées aux mécanismes en jeu dans le développement du système nerveux.
Modèles de blessure traumatique
L’utilisation de modèles in vitro pour simuler une blessure traumatique a également évolué de façon significative au cours des dernières années [8]. La première technique consistait à déchirer ou à lacérer des cellules en culture avec une pointe, un poinçon ou un laser [9]. Mais, une autre technique très utilisée in vitro est celle de l’étirement des cellules, qui reproduit l’étendue et le taux de déformation du tissu survenant in vivo pendant une blessure [10]. Il est ainsi possible de repérer les cellules du système nerveux vulnérables à certains types de déformations mécaniques et d’évaluer si les mécanismes de blessures changent lorsque la déformation des cellules se fait par étirement, par compression ou par la force de cisaillements [11, 12].
Bien que ces modèles d’explants reproduisent très fidèlement l’environnement tridimensionnel du tissu nerveux in vivo, ils présentent l’inconvénient majeur de ne permettre aucune modification de leur contenu cellulaire et de leur organisation. On voudrait ainsi pouvoir étudier le comportement des neurones en présence ou en l’absence de certains de leurs partenaires ; plus précisément, on voudrait pouvoir faire des combinaisons de cellules nerveuses provenant de patients atteints de maladies neurodégénératives avec celles issues de sujets sains pour déterminer l’origine cellulaire de la maladie.
La seule façon de pouvoir pleinement choisir le type de cellules, leur origine (malade ou saine), leur nombre et leur organisation dans un environnement en trois dimensions mimant le système nerveux est de reconstruire ce tissu le plus complètement possible en ayant le choix de chacun de ses composants.
Le génie tissulaire appliqué au système nerveux
La principale difficulté pour modéliser le système nerveux est précisément son aspect tridimensionnel, qui exige de cultiver les cellules dans une architecture 3D.
Notre équipe a acquis une longue expérience dans la reconstruction d’organes en trois dimensions. L’architecture que nous utilisons souvent pour amener les cellules à former un tissu est une éponge de collagène. Il s’agit d’un biomatériau formé principalement de collagène bovin de type I et III, stabilisé par des liaisons ioniques assurées par du chitosane (un polysaccharide d’origine marine, biodégradable, biocompatible et bioactif) dont les dimensions peuvent être ajustées à volonté, mais que l’on utilise en général coulé directement dans des plaques de culture 12 puits, avec un diamètre de 22 mm et une épaisseur de 1,5 mm. Nous avons montré que cette éponge conduisait à la formation d’un tissu conjonctif de grande qualité lorsqu’on y cultivait des fibroblastes [13]. En revanche, son utilisation directement avec des cellules nerveuses (neurones matures et cellules gliales) s’est révélée infructueuse, car celles-ci ne sont ni des cellules ayant la capacité de migrer, dans le cas des neurones, ni susceptibles de sécréter une matrice extracellulaire en abondance, deux propriétés essentielles au remodelage du biomatériau.
Nous avons donc choisi une autre stratégie, celle qui consiste à reconstruire d’abord un tissu conjonctif avec une éponge ensemencée avec des fibroblastes et à ensemencer ensuite à sa surface les cellules nerveuses pour y favoriser la migration axonale des neurones. L’éponge représente alors le tissu conjonctif à travers lequel les nerfs migrent jusqu’à atteindre leur cible, un muscle, par exemple. La couche de cellules nerveuses à la surface de l’éponge mime alors la moelle épinière. Ce principe a été appliqué à l’étude de la migration axonale des neurones sensoriels et des neurones moteurs [14, 15].
Les fibroblastes servant à la reconstruction du tissu conjonctif sont isolés à partir de la peau. Les cellules de Schwann, elles, sont reconstituées à partir de nerfs sciatiques de souris adultes. Les neurones moteurs, neurones sensoriels, microglies et astrocytes proviennent de moelles épinières d’embryons de souris ou de ganglions nerveux rachidiens [16].
L’éponge est ensemencée avec les fibroblastes (avec ou sans cellules de Schwann) pour reconstruire un tissu conjonctif, puis cultivée deux semaines. Pendant la troisième semaine, l’éponge est placée en culture à l’interface air-liquide pour établir un gradient de facteurs de croissance stimulant la migration axonale à travers le tissu. Les neurones sont ensuite ensemencés à la surface de l’éponge pour deux à trois semaines de culture supplémentaire (Figure 1, Figure 2A).
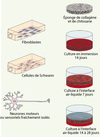 |
Figure 1. Le modèle d’étude de la migration axonale des neurones moteurs en trois dimensions. Le modèle est d’abord composé d’une éponge de collagène-chitosane, qui sert d’architecture 3D, dans laquelle sont ensemencés des fibroblastes avec ou sans cellules de Schwann. Après 3 semaines de maturation, les neurones moteurs sont déposés à la surface de l’éponge et cultivés 2 à 3 semaines supplémentaires à l’interface air-liquide pour favoriser la migration axonale vers le bas de l’éponge grâce à la présence des facteurs neurotrophiques dans le milieu de culture. |
 |
Figure 2. Évaluation du modèle en 3D. A. Aspect histologique de la couche de neurones moteurs déposée au-dessus de l’éponge colonisée par les fibroblastes, qui ont sécrété de grandes quantités de matrices extracellulaires (en bleu).B. Migration à travers l’éponge d’un grand nombre d’axones exprimant le neurofilament M (en vert).C, D. Après 28 jours de maturation des neurones moteurs en présence de cellules de Schwann, la formation de gaines de myéline autour des axones migrant à travers l’éponge est observée en microscopie électronique à transmission. N : neurone, m : myéline. D est un agrandissement de C. Barres en A : 60 µm, en B : 100 µm, en C : 2 µm et en D : 0,2 µm (modifié de [14] avec l’autorisation de Wiley Interscience). |
En présence de facteurs neurotrophiques, BDNF (brain-derived neurotrophic factor), NT3 (neurotrophin 3), CNTF (ciliary neurotrophic factor), GDNF (glial cell-derived neurotrophic factor) pour les neurones moteurs, NGF (nerve growth factor) pour les neurones sensoriels, une migration axonale est observée sur une distance de près de 1 mm (visualisée par un marquage en immunofluorescence du neurofilament M, un marqueur des neurones) (Figure 2B), qu’il s’agisse de neurones sensoriels ou moteurs [14, 15]. De plus, ce modèle tridimensionnel permet la maturation des axones émis par les neurones moteurs, puisque le tiers de ceux-ci exprime, en plus du neurofilament M présent dans toutes les fibres, le neurofilament H, un marqueur d’une différenciation poussée de ces axones. Enfin, en présence de cellules de Schwann, nous avons non seulement démontré que ces cellules migraient spontanément pour se placer le long des axones, mais, en plus, qu’elles formaient des gaines de myéline autour d’eux après 28 jours de culture (Figure 2C, D) [14]. C’est la première fois que la formation de gaines de myéline in vitro autour d’axones de neurones moteurs est démontrée. Ce résultat ouvre des perspectives très encourageantes pour l’étude des maladies neurodégénératives dans lesquelles la gaine de myéline est atteinte (syndrome de Guillain-Barré par exemple).
Un autre aspect innovant de ce modèle tient à la possibilité unique de reproduire in vitro à l’intérieur de l’éponge un réseau bien structuré de pseudo-capillaires. Ces capillaires forment une lumière étanche, bien qu’ils ne contiennent pas de sang comme in vivo [17]. Il est donc possible de combiner, dans notre modèle, à la fois un réseau de pseudo-capillaires et un réseau nerveux et, donc, d’étudier les interactions croisées entre ces deux systèmes. En effet, il existe de nombreuses similitudes biologiques et moléculaires entre les systèmes nerveux et vasculaire. Il a été ainsi démontré que l’innervation sensorielle détermine l’alignement des petits vaisseaux sanguins de la peau et leur différenciation en artériole ou en veinule, entre autres, par l’intermédiaire d’une sécrétion de VEGF (vascular endothelial growth factor) [18]. Des résultats préliminaires nous ont permis d’établir que, dans ce modèle, les nerfs sensitifs produisent un effet angiogénique.
Ce modèle présente donc une grande flexibilité, puisqu’il peut être constitué, au choix, de différents types de neurones, de différents types de cellules gliales (astrocytes, oligodendrocytes, microglies, cellules de Schwann) et d’autres types cellulaires comme les cellules endothéliales ou musculaires, provenant de souris sauvages ou de souris transgéniques ou ayant un phénotype pathologique. De plus, comme il peut être maintenu en culture pendant plus d’un mois en présence des neurones, il peut être utilisé pour tester l’effet de différents types de molécules sur la migration axonale, la myélinisation ou la cytotoxicité neuronale.
Le principal inconvénient de ce modèle est sa complexité, car il requiert une bonne maîtrise des techniques de génie tissulaire pour reconstruire un tissu en trois dimensions. De plus, la contrainte inhérente à tous les modèles in vitro utilisant des neurones est l’obligation d’obtenir des cellules fraîchement isolées à chaque expérience. Cet impératif se traduit par une quantité de travail et un coût importants, qui en rendent difficile la commercialisation, à moins d’utiliser des lignées cellulaires transformées. D’autre part, l’imagerie du réseau nerveux dans un tissu en trois dimensions est nécessairement plus compliquée à réaliser que dans un réseau monocouche, et exige d’utiliser la microscopie confocale. Enfin, le mélange de plusieurs types cellulaires rend difficiles les études d’expression des gènes.
Ce modèle pourra se révéler extrêmement performant dès lors qu’il pourra être constitué de cellules prélevées chez des patients atteints d’une maladie neurodégénérative. Des progrès récents dans le domaine des cellules souches permettent d’espérer pouvoir obtenir ces cellules par différenciation, à partir de précurseurs neuraux tirés d’une source de tissu accessible. Nous avons ainsi démontré qu’il était possible de différencier en neurones des précurseurs neuraux isolés à partir de peau normale humaine [19] et nous travaillons également à les différencier en cellules de Schwann. Ces travaux ouvrent la perspective de développer un modèle entièrement humain, constitué de cellules neurales différenciées à partir d’une petite biopsie de peau prélevée chez des patients souffrant, par exemple, de la maladie de Parkinson ou de la sclérose latérale amyotrophique. En déterminant quelle combinaison contenant le moins de cellules malades par rapport aux cellules saines induit la dégénérescence nerveuse, il serait possible de mieux cerner l’origine de la maladie, ainsi que son processus évolutif.
Conclusion
Bien que la culture des neurones et des cellules gliales in vitro se fasse de manière standard depuis de nombreuses années et que l’utilisation des tranches de tissus nerveux en culture organotypique constitue un excellent modèle préservant la structure tridimensionnelle du tissu, la reconstruction plus ou moins complète du système nerveux par des techniques de génie tissulaire représente une voie prometteuse pour aider à comprendre sa physiologie et les pathologies qui y sont associées. Le principal avantage des tissus reconstruits est de permettre de choisir chacun des éléments qui les constituent, un avantage qui prend tout son sens avec la perspective d’utiliser des précurseurs humains adultes prélevés chez des patients, comme source de neurones et de cellules gliales. Dès lors, la modélisation in vitro des maladies neurodégénératives avec un système de culture mimant la condition physiologique du tissu d’origine et composé de cellules d’un patient devient possible. De tels modèles devraient fournir des outils précieux à la communauté scientifique pour mieux comprendre l’origine et le développement de ces maladies souvent encore incurables.
Remerciements
Nos travaux sont financés par la Muscular Dystrophy Association (www.mda.org) et par les Instituts de recherche en santé du Canada (IRSC).
Références
- Varghese K, Das M, Bhargava N, et al. Regeneration and characterization of adult mouse hippocampal neurons in a defined in vitro system.J Neurosci Methods 2009; 177 : 51–9. [Google Scholar]
- Viviani B, Corsini E, Binaglia M, et al. Reactive oxygen species generated by glia are responsible for neuron death induced by human immunodeficiency virus-glycoprotein 120 in vitro. Neuroscience 2001; 107 : 51–8. [Google Scholar]
- Polazzi E, Gianni T, Contestabile A. Microglial cells protect cerebellar granule neurons from apoptosis: evidence for reciprocal signaling. Glia 2001; 36 : 271–80. [Google Scholar]
- Hao HN, Zhao J, Thomas RL, et al. Fetal human hematopoietic stem cells can differentiate sequentially into neural stem cells and then astrocytes in vitro. J Hematother Stem Cell Res 2003; 12 : 23–32. [Google Scholar]
- Gahwiler BH. Organotypic monolayer cultures of nervous tissue.J Neurosci Methods 1981; 4 : 329–42. [Google Scholar]
- Llano I, Marty A, Johnson JW, et al. Patch-clamp recording of amino acid-activated responses in organotypic slice cultures. Proc Natl Acad Sci USA 1988; 85 : 3221–5. [Google Scholar]
- McKinney RA, Luthi A, Bandtlow CE, Gahwiler BH, Thompson SM. Selective glutamate receptor antagonists can induce or prevent axonal sprouting in rat hippocampal slice cultures. Proc Natl Acad Sci USA 1999;96 : 11631–6. [Google Scholar]
- Morrison B 3rd, Saatman KE, Meaney DF, McIntosh TK. In vitro central nervous system models of mechanically induced trauma: a review.J Neurotrauma 1998; 15 : 911–28. [Google Scholar]
- Regan RF, Choi DW. The effect of NMDA, AMPA/kainate, and calcium channel antagonists on traumatic cortical neuronal injury in culture. Brain Res 1994; 633 : 236–42. [Google Scholar]
- Meaney DF, Smith DH, Shreiber DI, et al. Biomechanical analysis of experimental diffuse axonal injury. J Neurotrauma 1995; 12 : 689–94. [Google Scholar]
- Geddes-Klein DM, Schiffman KB, Meaney DF. Mechanisms and consequences of neuronal stretch injury in vitro differ with the model of trauma.J Neurotrauma 2006; 2 3: 193–204. [Google Scholar]
- LaPlaca MC, Lee VM, Thibault LE. An in vitro model of traumatic neuronal injury: loading rate-dependent changes in acute cytosolic calcium and lactate dehydrogenase release. J Neurotrauma 1997; 14 : 355–68. [Google Scholar]
- Berthod F, Germain L, Guignard R, et al. Differential expression of collagen XII and XIV in human skin and in reconstructed skin. J Invest Dermatol 1997; 108 : 737–42. [Google Scholar]
- Gingras M, Beaulieu MM, Gagnon V, et al. In vitro study of axonal migration and myelination of motor neurons in a three-dimensional tissue-engineered model. Glia 2008; 56 : 354–64. [Google Scholar]
- Gingras M, Bergeron J, Dery J, et al. In vitro development of a tissue-engineered model of peripheral nerve regeneration to study neurite growth. FASEB J 2003; 17 : 2124–6. [Google Scholar]
- Gingras M, Gagnon V, Minotti S, et al. Optimized protocols for isolation of primary motor neurons, astrocytes and microglia from embryonic mouse spinal cord. J Neurosci Methods 2007; 163 : 111–8. [Google Scholar]
- Berthod F, Germain L, Tremblay N, Auger FA. Extracellular matrix deposition by fibroblasts is necessary to promote capillary-like tube formation in vitro. J Cell Physiol 2006; 207 : 491–8. [Google Scholar]
- Mukouyama YS, Shin D, Britsch S, et al. Sensory nerves determine the pattern of arterial differentiation and blood vessel branching in the skin. Cell 2002; 109 : 693–705. [Google Scholar]
- Gingras M, Champigny MF, Berthod F. Differentiation of human adult skin-derived neuronal precursors into mature neurons. J Cell Physiol 2007; 210 : 498–506. [Google Scholar]
Liste des figures
|
Figure 1. Le modèle d’étude de la migration axonale des neurones moteurs en trois dimensions. Le modèle est d’abord composé d’une éponge de collagène-chitosane, qui sert d’architecture 3D, dans laquelle sont ensemencés des fibroblastes avec ou sans cellules de Schwann. Après 3 semaines de maturation, les neurones moteurs sont déposés à la surface de l’éponge et cultivés 2 à 3 semaines supplémentaires à l’interface air-liquide pour favoriser la migration axonale vers le bas de l’éponge grâce à la présence des facteurs neurotrophiques dans le milieu de culture. |
| Dans le texte | |
|
Figure 2. Évaluation du modèle en 3D. A. Aspect histologique de la couche de neurones moteurs déposée au-dessus de l’éponge colonisée par les fibroblastes, qui ont sécrété de grandes quantités de matrices extracellulaires (en bleu).B. Migration à travers l’éponge d’un grand nombre d’axones exprimant le neurofilament M (en vert).C, D. Après 28 jours de maturation des neurones moteurs en présence de cellules de Schwann, la formation de gaines de myéline autour des axones migrant à travers l’éponge est observée en microscopie électronique à transmission. N : neurone, m : myéline. D est un agrandissement de C. Barres en A : 60 µm, en B : 100 µm, en C : 2 µm et en D : 0,2 µm (modifié de [14] avec l’autorisation de Wiley Interscience). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.