")
")
| Issue |
Med Sci (Paris)
Volume 24, Number 8-9, Août-Septembre 2008
|
|
|---|---|---|
| Page(s) | 725 - 730 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/20082489725 | |
| Published online | 15 août 2008 | |
Le double jeu de l’épigénétique
Cible et acteur du cancer
Master and servant: epigenetic deregulations as a cause and a consequence of cancer
1
New England Biolabs, 240 County Road, Ipswich, MA 01938, États-Unis.
2
CNRS UMR 218, Institut Curie, 26, rue d’Ulm, 75248 Paris Cedex 05, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Le mot « épigénétique » désigne les phénomènes transmis héréditairement, et qui modulent l’activité du génome sans changer sa séquence. Nous abordons le lien entre épigénétique et cancer par ces questions : quels sont les processus épigénétiques et comment sont-ils altérés dans les cancers ? Comment l’épigénétique se combinet- elle à la génétique pour entraîner un processus cancéreux ? Quelles sont les conséquences pour le diagnostic et la thérapie ? Quels liens entre épigénétique et environnement ? Nous arriverons à la conclusion que les changements épigénétiques sont à la fois cibles et acteurs dans le processus tumoral
Abstract
The processes that affect the activity of the genome in a heritable manner without changing its sequence are called epigenetic. Here we review the modes of epigenetic gene regulation, and describe their alterations in cancer. We show how these mechanisms are interdependent, and how they intersect with genetic mutations. We argue that epigenetic abnormalities can occur both as a cause, and as a consequence of cancer. Indeed, oncogenic transformation can deeply alter the epigenetic information contained in the pattern of DNA methylation or histone tail modification. Conversely, epigenetic dysfunctions can drive cellular transformation. We then touch on some practical consequences of the prominence of epigenetic alterations in cancer : increasing knowledge of this field has allowed the development of a new generation of diagnostic tools and therapeutic avenues. Finally we point out that epigenetic phenomena may act as sensors that link environmental conditions to cancer
© 2008 médecine/sciences - Inserm / SRMS
Cancer et épigénétique
Le cancer est un problème de santé publique majeur : selon l’Organisation Mondiale de la Santé, cette maladie a tué 7,6 millions de personnes dans le monde en 2005 (http://www.who.int/fr). Le terme générique « cancer » recouvre différentes pathologies : il existe environ 200 types de tumeurs pouvant affecter tous les tissus du corps. Ces affections résultent toutes de l’acquisition et du maintien par les cellules de caractères anormaux : l’indépendance vis-à-vis des signaux de croissance, la résistance aux signaux inhibant la croissance, la résistance à la mort cellulaire programmée ou apoptose, l’acquisition d’un potentiel réplicatif illimité, la capacité à susciter la genèse de vaisseaux sanguins ou angiogenèse, et la capacité à former des métastases. Ces six propriétés, chacune plus ou moins développée selon les tumeurs, résultent de l’altération de l’expression ou de la séquence d’oncogènes et de gènes suppresseurs de tumeurs [1].
Le génome contient plus de 20 000 gènes codant des protéines, mais chaque type cellulaire n’en exprime qu’un sous-ensemble. Ainsi, certains gènes exprimés dans les neurones sont silencieux dans les cellules musculaires, et vice versa. Différentes régulations contrôlant l’expression des gènes sont de nature épigénétique, c’est-à-dire qu’elles modulent l’activité du génome sans modifier sa séquence et se transmettent à leur descendance [2, 31] (Figure 1).
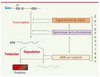 |
Figure 1. Les mécanismes épigénétiques agissant sur l’expression des gènes. Les différents mécanismes épigénétiques peuvent agir sur la transcription des gènes, mais aussi à des étapes ultérieures, dont celle de la traduction des ARN. Ces processus sont physiquement et fonctionnellement reliés au sein du noyau. |
Dans cette revue, nous discutons des liens entre épigénétique et cancer. Une sélection a été nécessaire et certains phénomènes, dont l’organisation des domaines nucléaires, ne seront pas exposés. Nous traiterons principalement des processus épigénétiques agissant sur la chromatine - méthylation de l’ADN, dynamique des histones et des protéines non histones - ainsi que du rôle des ARN non codants.
La méthylation de l’ADN, ses acteurs et ses interprètes
Les cytosines peuvent subir une modification chimique, la méthylation. Elle ne peut avoir lieu que lorsque la cytosine est suivie d’une guanine, on parle de dinucléotide CpG. La méthylation est mise en place par les méthyltransférases de l’ADN ayant une activité dite de novo, DNMT1, DMNT3a et DNMT3b [32]. Cette marque est ensuite maintenue au cours des divisions cellulaires par l’enzyme de maintien, DNMT1.
Dans les cellules saines, la méthylation touche principalement les séquences d’ADN répétées et les gènes soumis à l’empreinte [3]. En revanche, les îlots riches en CpG présents dans les promoteurs des gènes sont pour la plupart non méthylés. Les cellules cancéreuses ont un taux global de méthylation du génome inférieur à celui de cellules saines, avec un patron de méthylation radicalement modifié (→).
(→) Voir l’article de M. Weber, page 731 de ce numéro
Les anomalies de méthylation de l’ADN participent à la transformation par quatre voies (Figure 2) : (1) Les cytosines méthylées peuvent subir spontanément une déamination qui les transforme en thymine, causant un mésappariement T-G, et une possible transition C->T. On estime que plus de 35 % des mutations ponctuelles sont dues à ce phénomène [4] ; (2) les gènes suppresseurs de tumeurs dont le promoteur contient un îlot CpG peuvent être inactivés par la méthylation de ce dernier (Figure 3). En effet cette hyperméthylation attire des protéines qui reconnaissent l’ADN méthylé, et recrutent divers corépresseurs agissant notamment sur l’organisation de la chromatine [5, 6] ; (3) les gènes soumis à l’empreinte parentale peuvent voir leur expression perturbée, ce qui peut contribuer à la transformation (→) ; et (4), la déméthylation des séquences répétées diminue la stabilité du génome [7] et favorise la réactivation d’éléments mobiles, les transposons. Les liens mécanistiques reliant la transformation cellulaire aux anomalies de la méthylation de l’ADN commencent à être déchiffrés [8].
 |
Figure 2. Différents mécanismes épigénétiques associés au cancer. Les symboles utilisés sont les même que ceux de la Figure 3. |
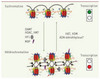 |
Figure 3. Conversion épigénétique entre euchromatine et hétérochromatine. Les modifications épigénétiques agissent de concert pour permettre la transition réversible de l’euchromatine, permissive pour la transcription, à l’hétérochromatine, qui est généralement répressive pour la transcription [33]. Sur ce schéma sont représentés : (1) l’ADN (en noir) (2) la méthylation de l’ADN : les méthylcytosines sont symbolisées par des points roses ; (3) les nucléosomes, autour desquels l’ADN est enroulé. Ils sont constitué de 2 copies de chacune des 4 histones canoniques : H2A (en bleu), H2B (en vert), H3 (en rouge) et H4 (en jaune) ; (4) les modifications post-traductionnelles des histones. Dans un but de simplification, nous avons représenté par un seul symbole (carrés) les différentes marques typiques de l’euchromatine, et par un autre (triangle) celles qui sont caractéristiques de l’hétérochromatine ; (5) les variants des histones (bleu clair signalé par une flèche rouge). Tous les variants ne sont pas mentionnés ici. Les protéines HMG et les ARN non codants n’ont pas été représentés. |
(→) Voir l’article de A. Henckel et R. Feil, page 747 de ce numéro
Les protéines liant spécifiquement l’ADN méthylé sont l’interface entre méthylation de l’ADN et chromatine. À ce jour 3 familles sont connues : (1) la famille des protéines à MBD (methyl-binding domain) [9] ; (2) la famille de protéines à doigts de Zinc, contenant Kaiso, ZBTB4, et ZBTB38 [10] ; et (3) un groupe de deux protéines à domaine SRA (SET and RING-finger associated), UHRF1 et UHRF2 [11]. Les protéines MBD, ainsi que celles apparentées à Kaiso, sont des répresseurs transcriptionnels. Elles peuvent agir comme oncogènes, en réprimant l’expression de gènes suppresseurs de tumeur dont le promoteur a été méthylé. Ainsi, la délétion de MBD2, ou celle de Kaiso, freine la croissance des tumeurs intestinales chez les souris de fond génétique APCmin, qui présentent une activation de la voie Wnt1. Ces protéines pourraient donc devenir des cibles thérapeutiques [12].
La dynamique des histones et autres protéines chromatiniennes
L’ADN est enroulé autour des nucléosomes, qui sont constitués de protéines, les histones (→). Les histones peuvent être chimiquement modifiées par différentes enzymes : histone acétyl-transférases (HAT) et histone déacétylases (HDAC) ; histone méthytranférases (HMT) et histone déméthylases (HDM) notamment [2]. Ces modifications réversibles régulent le recrutement de cofacteurs sur les nucléosomes et modifient ainsi l’activité de la chromatine. Certaines modifications sont caractéristiques de l’hétérochromatine, d’autres de l’euchromatine, et un « code des histones » a été proposé [13].
(→) Voir l’article de A. Bertin et S. Mangenot, page 715 de ce numéro
Dans les cellules cancéreuses, il existe des changements dans les modifications des histones. Ces changements peuvent être subtils, et ne toucher que la région promotrice de certains gènes [14]. Ils peuvent aussi être plus massifs : dans les cellules transformées, l’acétylation de la lysine 16 et la triméthylation de la lysine 20 de l’histone H4 peuvent être perdues au niveau de nombreuses séquences répétées [15]. La dérégulation des modifications des histones est souvent causée par des anomalies touchant les enzymes correspondantes. Ces protéines peuvent être sous- ou sur-exprimées, mutées, ou fusionnées à une autre protéine via une translocation [16–18].
En plus de leur rôle dans la transcription, les histones interviennent dans la détection et la réparation des dommages subis par l’ADN. Le variant H2A.X est particulièrement important : la délétion d’un seul de ses allèles compromet l’intégrité du génome et augmente la susceptibilité au cancer [19].
Enfin, d’autres protéines de la chromatine peuvent aussi être dérégulées. Par exemple, des protéines de la famille HMG (High mobility group) sont surexprimées dans certains cancers [20]. Les protéines HP1 interprètent les modifications post-traductionnelles des histones2. La sous-expression de la protéine HP1α est associée avec le cancer du sein métastatique [21]. Dans ces deux cas, il est probable que les anomalies chromatiniennes accélèrent la transformation en perturbant l’expression des gènes et/ou en fragilisant certaines régions du génome, favorisant ainsi l’apparition d’anomalies chromosomiques supplémentaires.
Les ARN non codants
La perturbation de la machinerie de génération des petits ARN contribue à la transformation cellulaire [22] sans doute pour plusieurs raisons. Les microARN modulent l’expression des gènes en agissant sur la traduction ou la dégradation des ARN messagers (Figure 1). Certains microARN [23] sont des suppresseurs de tumeur ; d’autres, en étant anormalement exprimés, ou bien mutés, sont au contraire oncogéniques. D’autres ARN non codants, issus des régions centromériques, jouent un rôle dans la ségrégation des chromosomes et sont donc importants pour la stabilité du génome [24].
Les régulateurs épigénétiques forment un réseau complexe
Les différents phénomènes épigénétiques sont fortement connectés. Il existe de multiples cas d’interaction fonctionnelle entre deux phénomènes donnés (Figure 1). Ainsi l’ADN-méthyltransférase DNMT1 peut agir en synergie avec les ADN- méthyltransférases DNMT3a et b, les histones méthyltransférases Suv39h1 et G9a et avec l’histone déacétylase HDAC2, mais elle peut aussi interagir avec les complexes de remodelage des histones [25].
L’existence de ces multiples boucles de régulation rend difficile l’établissement des liens de cause à effet. Par exemple : la méthylation des gènes cause-t-elle leur inactivation transcriptionnelle, ou la méthylation résulte-t-elle au contraire du manque d’activité de gènes déjà réprimés ? [26]. Mais, l’intégration des phénomènes épigénétiques en un réseau complexe présente paradoxalement un espoir thérapeutique : il signifie qu’un point de régulation donné peut être modifié directement, mais aussi indirectement, en agissant à un autre niveau du système. Rappelons que, à l’inverse des mutations, les modifications épigénétiques sont en théorie réversibles.
Vers des moyens de diagnostic et des thérapies « épigénétiques » ?
Le diagnostic précoce du cancer améliore fortement les chances de survie. Les altérations épigénétiques des cellules cancéreuses, notamment l’hyperméthylation de promoteurs, peuvent être détectées de façon simple et non invasive, à partir de cellules collectées dans les fluides corporels. Cette détection permet d’intervenir rapidement, mais aussi de choisir un traitement [27].
D’autre part, les acteurs de l’épigénétique sont des cibles thérapeutiques. La 5-aza-déoxycytidine, un inhibiteur des DNMT, est efficace contre les myélodysplasies [2]. Une autre approche médicamenteuse vise les HDAC (→) [34].
(→) Voir l’article de D. Mottet et V. Castronovo, page 742 de ce numéro
Des effets synergiques des inhibiteurs de HDAC et de DNMT sont d’ailleurs observés [28]. Enfin, d’autres espoirs thérapeutiques reposent sur l’utilisation de miRNA dirigés contre les oncogènes [29]. Par ailleurs, les anomalies épigénétiques des cellules cancéreuses causent l’expression d’antigènes normalement absents des cellules somatiques [35] (→).
(→) Voir l’article de S. Rousseaux et al., page 735 de ce numéro
La vaccination contre ces antigènes pourrait être un autre moyen de cibler les cellules tumorales.
Environnement et cancer : l’épigénétique comme senseur ?
Le risque de cancer du sein est six fois plus élevé aux Etats-unis qu’en Chine [1]. L’explication n’est pas génétique : les enfants d’immigrés chinois vivant aux États-Unis présentent un risque égal à celui du reste de la population américaine. L’environnement joue donc un rôle clair dans la fréquence et la nature des cancers touchant une population. De fait, chaque population est exposée à une combinaison de conditions physiques (par exemple, le rayonnement solaire) ou chimiques (notamment par l’alimentation) qui lui sont propres, et qui expliquent en partie le patron de tumeurs observé. Ces facteurs peuvent bien sûr agir au niveau génétique, en favorisant ou en limitant certaines mutations. Mais ils peuvent aussi agir au niveau épigénétique (Figure 4) [30].
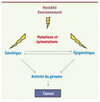 |
Figure 4. Les facteurs de risques du cancer. Les mutations, qui affectent le matériel génétique, peuvent être héritées de façon familiale, ou être dues à des mutagènes environnementaux (rayons UV, fumée de cigarette, alimentation). De façon similaire, il existe aussi des anomalies touchant l’information épigénétique, qu’on appelle « épimutations ». Des mutations génétiques peuvent affecter la machinerie qui maintient l’information épigénétique. Réciproquement, des défauts épigénétiques peuvent fragiliser le matériel génétique. Cette combinaison d’anomalies résulte in fine en une modification de l’activité du génome conférant aux cellules des caractères anormaux. Ceux qui confèrent des avantages adaptatifs sont sélectionnés dans les tumeurs. |
Ainsi, chez la souris, un transposon est intégré à proximité du promoteur du gène Agouti. Si ce transposon est méthylé, l’expression d’Agouti est régulée correctement. Mais, s’il est déméthylé, le transposon active anormalement l’expression d’Agouti. Il s’ensuit un changement de couleur du pelage de l’animal, car Agouti agit sur la pigmentation. Mais d’autres effets sont plus graves : les souris deviennent obèses et présentent une fréquence accrue de cancers. Cet exemple présente deux caractéristiques spectaculaires. D’abord, la méthylation du transposon, et donc la susceptibilité aux tumeurs, est directement liée à la nutrition. En effet, un régime riche en donneurs de méthyle (folate, vitamine B12…) favorise la méthylation du transposon, et inhibe son action. Deuxièmement, l’effet est transgénérationnel : l’alimentation de la mère détermine le phénotype des adultes des générations suivantes, par le biais de la méthylation de l’ADN. Il semble d’ailleurs que ce mécanisme puisse s’appliquer aussi à d’autres types d’effets transgénérationnels, dont l’obésité.
L’épigénétique se combine à la génétique pour la transformation cellulaire
Une des causes de la tumorigenèse est l’altération des oncogènes et des suppresseurs de tumeurs au niveau génétique, par mutation ponctuelle, délétion, ou translocation. Les phénomènes épigénétiques peuvent se combiner aux anomalies génétiques de trois manières.
L’inactivation des suppresseurs de tumeurs peut se faire par une combinaison d’anomalies génétiques et épigénétiques. Ainsi, la perte de p53 peut résulter de la mutation d’un allèle, et de la méthylation de l’autre allèle [5, 6].
Les paramètres épigénétiques peuvent augmenter la sensibilité du matériel génétique aux mutations. Ainsi, les cytosines méthylées sont intrinsèquement mutagènes (voir plus haut), mais aussi plus sensibles que les cytosines non modifiées aux rayons UV et à la formation d’adduits avec le benzopyrène de la fumée de cigarette. L’empaquetage de l’ADN sous forme de chromatine modifie également son accessibilité aux mutagènes chimiques, ainsi que la capacité de la cellule à réparer les lésions génétiques qu’elle a subies.
Des altérations génétiques peuvent toucher des effecteurs épigénétiques. Ainsi la transformation de cellules par l’oncogène K-ras mobilise la machinerie de méthylation de l’ADN, de remodelage de la chromatine, et de modification des histones [8]. Par ailleurs, de nombreuses enzymes modifiant les histones, ou remodelant les nucléosomes, subissent des translocations, des mutations, ou des délétions, dans les cancers [5, 6].
Conclusion
La compréhension des mécanismes de développement du cancer a beaucoup progressé au cours des décennies 1980 et 1990. Les travaux réalisés ont permis de cataloguer de nombreux oncogènes et gènes suppresseurs de tumeurs, et de montrer qu’ils sont la cible d’anomalies génétiques qui mènent à la transformation cellulaire. Plus récemment, il est apparu que les différents processus épigénétiques sont également des acteurs importants de ces transformations, qui peuvent intervenir seuls, ou en combinaison avec des perturbations génétiques. Cette situation est complexe à deux titres. Premièrement, les mécanismes épigénétiques forment un réseau intriqué comprenant des boucles d’amplification et de rétro-action. Il est difficile d’y étudier un phénomène sans en affecter d’autres. Deuxièmement les liens entre génétique et épigénétique sont également abondants et bidirectionnels. Les liens de cause à effet peuvent notamment être ardus à établir.
La modélisation et l’analyse de tels réseaux sont un des buts de cette nouvelle discipline qu’est la biologie des systèmes. La conception d’un modèle suffisamment fidèle à la réalité aurait plusieurs avantages : elle donnerait une vision d’ensemble de ces mécanismes, faciliterait l’identification des nœuds défaillants et le choix d’interventions compensatrices. Par analogie, les réseaux protéine-ADN-ARN existant à un temps donné peuvent être comparés à un circuit électrique. A ce jour, les composants du circuit sont loin d’avoir tous été caractérisés. Le programme génétique pourrait être alors comparé au logiciel qui gouverne le circuit. Ce programme est difficile à décoder car il est interactif et réagit aux variations de l’environnement.
Sans nier les difficultés, nous pouvons être optimistes. Une meilleure connaissance des mécanismes épigénétiques, outre son intérêt fondamental, devrait en effet permettre à l’avenir l’amélioration du diagnostic et de la thérapie du cancer.
Les souris Min porteuses d’une mutation dans le gène APC (adenomatous polyposis coli), un intermédiaire de la voie de signalisation Wnt, développent une polypose très semblable à la maladie humaine et représentent un bon modèle animal de progression tumorale.
Les protéines HP1 (heterochromatin protein 1) sont une des composantes essentielles de l’hétérochromatine péricentrique. La présence d’un domaine de dimérisation dans sa partie carboxy-terminale permet à HP1 de fixer simultanément deux molécules d’H3 méthylées sur la Lys9 de manière intra- ou inter-nucléosomique [copié de 34].
Remerciements
Le laboratoire de PAD est financé par l’Association pour la Recherche contre le Cancer, la Ligue Nationale contre le Cancer, l’Institut National du Cancer, et le CNRS. SL remercie le Dr Richard J. Roberts et New England Biolabs pour leur soutien, et le Dr Pierre-Olivier Estève pour ses commentaires sur le manuscrit.
Références
- Weinberg RA. The biology of cancer. New York : Garland Science 2006. [Google Scholar]
- Allis CD, Jenuwein T, Reinberg D, et al. Epigenetics. New York : Cold Spring Harbor Laboratory Press, 2006. [Google Scholar]
- Gabory A, Dandolo D. Épigénétique et développement : l’empreinte parentale. Med Sci (Paris) 2005; 21 : 390–5. [Google Scholar]
- Rideout WM, Coetzee GA, Olumi AF, et al. 5-Methylcytosine as an endogenous mutagen in the human LDL receptor and p53 genes. Science 1990; 249 : 1288–90. [Google Scholar]
- Klose RJ, Bird AP. Genomic DNA methylation : the mark and its mediators. Trends Biochem Sci 2006; 31 : 89–97. [Google Scholar]
- Deltour S, Chopin V, Leprince D. Modifications épigénétiques et cancer. Med Sci (Paris) 2005; 21 : 405–11. [Google Scholar]
- Chen RZ, Pettersson U, Beard C, et al. DNA hypomethylation leads to elevated mutation rates. Nature 1998; 395 : 89–93. [Google Scholar]
- Gazin C, Wajapeyee N, Gobeil S, et al. An elaborate pathway required for Ras-mediated epigenetic silencing. Nature 2007; 449 : 1073–7. [Google Scholar]
- Hendrich B, Tweedie S. The methyl-CpG binding domain and the evolving role of DNA methylation in animals. Trends Genet 2003; 19 : 269–77. [Google Scholar]
- Filion GJ, Zhenilo S, Salozhin S, et al. A family of human zinc finger proteins that bind methylated DNA and repress transcription. Mol Cell Biol 2006; 26 : 169–81. [Google Scholar]
- Bostick M, Kim JM, Esteve PO, et al. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science 2007; 317 : 1760–4. [Google Scholar]
- Sansom OJ, Maddison K, Clarke AR. Mechanisms of disease : methyl-binding domain proteins as potential therapeutic targets in cancer. Nat Clin Pract Oncol 2007; 4 : 305–15. [Google Scholar]
- Ray-Gallet D, Gerard A, Polo S, et al. Variations sur le thème du code histone. Med Sci (Paris) 2005; 21 : 384–9. [Google Scholar]
- Grignani F, De Matteis S, Nervi C, et al. Fusion proteins of the retinoic acid receptor-alpha recruit histone deacetylase in promyelocytic leukaemia. Nature 1998; 391 : 815–8. [Google Scholar]
- Fraga MF, Ballestar E, Villar-Garea A, et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet 2005; 37 : 391–400. [Google Scholar]
- Kuzmichev A, Margueron R, Vaquero A, et al. Composition and histone substrates of polycomb repressive group complexes change during cellular differentiation. Proc Natl Acad Sci USA 2005; 102 : 1859–64. [Google Scholar]
- Ropero S, Fraga MF, Ballestar E, et al. A truncating mutation of HDAC2 in human cancers confers resistance to histone deacetylase inhibition. Nat Genet 2006; 38 : 566–9. [Google Scholar]
- Lund AH, Van Lohuizen M. Epigenetics and cancer. Genes Dev 2004; 18 : 2315–35. [Google Scholar]
- Celeste AS, Difilippantonio MJ. H2AX haploinsufficiency modifies genomic stability and tumor susceptibility. Cell 2003; 114 : 371–83. [Google Scholar]
- Fusco A, Fedele M. Roles of HMGA proteins in cancer. Nat Rev Cancer 2007; 7 : 899–910. [Google Scholar]
- Kirschmann DA, Lininger RA, Gardner LM, et al. Down-regulation of HP1Hsalpha expression is associated with the metastatic phenotype in breast cancer. Cancer Res 2000; 60 : 3359–63. [Google Scholar]
- Kumar MS, Lu J, Mercer KL, et al. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat Genet 2007; 39 : 673–7. [Google Scholar]
- Esquela-Kerscher A, Slack FJ. Oncomirs : microRNAs with a role in cancer. Nat Rev Cancer 2006; 6 : 259–69. [Google Scholar]
- Bouzinba-Segard H, Guais A, Francastel C. Accumulation of small murine minor satellite transcripts leads to impaired centromeric architecture and function. Proc Natl Acad Sci USA 2006; 103 : 8709–14. [Google Scholar]
- Groth A, Rocha W, Verreault A, et al. Chromatin challenges during DNA replication and repair. Cell 2007; 128 : 721–33. [Google Scholar]
- Baylin S, Bestor TH.Altered methylation patterns in cancer cell genomes : cause or consequence ? Cancer Cell 2002; 1 : 299–305. [Google Scholar]
- Esteller M. Epigenetic gene silencing in cancer : the DNA hypermethylome. Hum Mol Genet 2007; 16 : R50–9. [Google Scholar]
- Gius D, Cui H, Bradbury CM, et al. Distinct effects on gene expression of chemical and genetic manipulation of the cancer epigenome revealed by a multimodality approach. Cancer Cell 2004; 6 : 361–71. [Google Scholar]
- Brummelkamp TR, Bernards R, Agami R. Stable suppression of tumorigenicity by virus-mediated RNA interference. Cancer Cell 2002; 2 : 243–7. [Google Scholar]
- Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet 2007; 8 : 253–62. [Google Scholar]
- Képès F. L’épigénétique comme aspect de la postgénomique. Med Sci (Paris) 2005; 21 : 371–6. [Google Scholar]
- Fuks F. Les méthyltransférases de l’ADN : du remodelage de la chromatine au cancer. Med Sci (Paris) 2003; 19 : 477–80. [Google Scholar]
- Verreault A. Hétérochromatine : un silence bien bruyant. Med Sci (Paris) 2003; 19 : 1181–2. [Google Scholar]
- Mottet D, Castronovo V. Les histones désacétylases : nouvelles cibles thérapeutiques anti-cancéreuses. Med Sci (Paris) 2008; 24 : 742–6. [Google Scholar]
- Rousseaux S, Reynoird N, Gaucher J, Khochbin S. L’intrusion des régulateurs de l’épigénome mâle dans les cellules somatiques cancéreuses. Med Sci (Paris) 2008; 24 : 735–41. [Google Scholar]
Liste des figures
|
Figure 1. Les mécanismes épigénétiques agissant sur l’expression des gènes. Les différents mécanismes épigénétiques peuvent agir sur la transcription des gènes, mais aussi à des étapes ultérieures, dont celle de la traduction des ARN. Ces processus sont physiquement et fonctionnellement reliés au sein du noyau. |
| Dans le texte | |
|
Figure 2. Différents mécanismes épigénétiques associés au cancer. Les symboles utilisés sont les même que ceux de la Figure 3. |
| Dans le texte | |
|
Figure 3. Conversion épigénétique entre euchromatine et hétérochromatine. Les modifications épigénétiques agissent de concert pour permettre la transition réversible de l’euchromatine, permissive pour la transcription, à l’hétérochromatine, qui est généralement répressive pour la transcription [33]. Sur ce schéma sont représentés : (1) l’ADN (en noir) (2) la méthylation de l’ADN : les méthylcytosines sont symbolisées par des points roses ; (3) les nucléosomes, autour desquels l’ADN est enroulé. Ils sont constitué de 2 copies de chacune des 4 histones canoniques : H2A (en bleu), H2B (en vert), H3 (en rouge) et H4 (en jaune) ; (4) les modifications post-traductionnelles des histones. Dans un but de simplification, nous avons représenté par un seul symbole (carrés) les différentes marques typiques de l’euchromatine, et par un autre (triangle) celles qui sont caractéristiques de l’hétérochromatine ; (5) les variants des histones (bleu clair signalé par une flèche rouge). Tous les variants ne sont pas mentionnés ici. Les protéines HMG et les ARN non codants n’ont pas été représentés. |
| Dans le texte | |
|
Figure 4. Les facteurs de risques du cancer. Les mutations, qui affectent le matériel génétique, peuvent être héritées de façon familiale, ou être dues à des mutagènes environnementaux (rayons UV, fumée de cigarette, alimentation). De façon similaire, il existe aussi des anomalies touchant l’information épigénétique, qu’on appelle « épimutations ». Des mutations génétiques peuvent affecter la machinerie qui maintient l’information épigénétique. Réciproquement, des défauts épigénétiques peuvent fragiliser le matériel génétique. Cette combinaison d’anomalies résulte in fine en une modification de l’activité du génome conférant aux cellules des caractères anormaux. Ceux qui confèrent des avantages adaptatifs sont sélectionnés dans les tumeurs. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.