")
")
| Issue |
Med Sci (Paris)
Volume 23, Number 11, Novembre 2007
|
|
|---|---|---|
| Page(s) | 910 - 916 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/20072311910 | |
| Published online | 15 November 2007 | |
Récents progrès dans l’étiopathogénie de la scoliose idiopathique de l’adolescent et nouveaux concepts moléculaires
Etiopathogenesis of adolescent idiopathic scoliosis and new molecular concepts
1
Centre de recherche, CHU Sainte-Justine, Laboratoires de génétique moléculaire et de biologie cellulaire et tissulaire des maladies musculo-squelettiques, 3175, chemin de la Côte-Ste- Catherine, Montréal (Québec), H3T 1C5 Canada
2
Centre de recherche CHU Sainte-Justine, Laboratoire de génétique moléculaire des maladies musculo-squelettiques, 3175, chemin de la Côte-Ste-Catherine, Montréal (Québec), H3T 1C5 Canada
3
Département de stomatologie, Faculté de médecine dentaire, Université de Montréal , Centre de Recherche, CHU Sainte-Justine, Laboratoire de Biologie cellulaire des maladies musculo-squelettiques, 3175, chemin de la Côte-Ste-Catherine, Montréal (Québec), H3T 1C5 Canada
4
Service d’orthopédie, CHU Sainte-Justine, Montréal ; Service d’orthopédie, Hôpital Shriners pour enfants de Montréal, Canada
5
Service d’orthopédie, Hôpital Shriners pour enfants de Montréal ; Service d’orthopédie Hôpital pour enfants de Montréal, Université McGill, Montréal, Canada
6
Département de biochimie, Faculté de médecine, Université de Montréal, Montréal, Canada
*
alain.moreau@recherche-ste-justine.qc.ca
La scoliose est la déformation la plus fréquente en orthopédie pédiatrique. Elle survient principalement à l’adolescence affectant les filles en plus grand nombre et avec le plus de sévérité. Nos travaux ont mis en évidence un défaut dans la transmission du signal de l’hormone mélatonine chez des patients gravement atteints de scoliose idiopathique de l’adolescent (SIA). Cette anomalie est causée par la phosphorylation de résidus sérine au niveau des protéines G inhibitrices (Gi) couplées aux récepteurs à mélatonine. Nous avons observé chez les patients SIA trois types de réponses anormales en présence de mélatonine ce qui suggère que ce défaut est attribuable à des gènes distincts en cause dans cette voie de signalisation. Cette découverte nous a permis de mettre au point un premier test de dépistage précoce à partir d’une simple prise de sang. Actuellement en cours de validation clinique au Canada, ce test devrait permettre l’identification des enfants porteurs du risque de développer une SIA
Abstract
Adolescent idiopathic scoliosis (AIS) is the most common form of scoliosis that affects a significant number of young teenagers, mainly females (0.2-6 % of the population). Historically, several hypothesis were postulated to explain the aetiology of AIS, including genetic factors, biochemical factors, mechanics, neurological, muscular factors and hormonal factors. The neuroendocrine hypothesis involving a melatonin deficiency as the source for AIS has generated great interest. This hypothesis stems from the fact that experimental pinealectomy in chicken, and more recently in rats maintained in a bipedal mode, produces a scoliosis. The biological relevance of melatonin in idiopathic scoliosis is controversial since no significant decrease in circulating melatonin level has been observed in a majority of studies. Analysis of melatonin signal transduction in musculoskeletal tissues of AIS patients demonstrated for the first time a defect occurring in a cell autonomous manner in different cell types isolated from AIS patients suffering of the most severe form of that disease. These results have led to a classification of AIS patients in three different functional groups depending on their response to melatonin, suggesting that the cause of AIS involves several genes. Molecular analysis showed that melatonin signaling dysfunction is triggered by an increased phosphorylation of Gi proteins inactivating their function. This discovery has led to development of a first scoliosis screening assay. This test, using blood sample, is currently in clinical validation process in Canada and could be used for screening children at high risk of developing AIS.
© 2007 médecine/sciences - Inserm / SRMS
La scoliose est la déformation la plus fréquente en orthopédie pédiatrique : elle se manifeste par une déformation structurale évolutive de la colonne vertébrale, de la cage thoracique et du bassin dans les trois plans de l’espace et dans le temps. Une large proportion soit 85 % des scolioses sont dites idiopathiques, cela signifie qu’on en ignore la cause ; les 15 % restants sont causées par des malformations congénitales, des maladies neuromusculaires ou associées à des pathologies osseuses ou à des pathologies des tissus conjonctifs. L’étiologie de la scoliose idiopathique de l’adolescent (SIA), la forme la plus commune de scoliose, n’est pas claire bien que la SIA soit une maladie affectant un nombre important de jeunes adolescents (0,2-6 % de la population), touchant principalement les filles en nombre et avec le plus de sévérité. Outre la déformation du rachis, cette maladie peut avoir des conséquences importantes sur la santé des patients et affecter des fonctions d’équilibre, vestibulaire et proprioceptive, altérer le tonus musculaire et susciter l’apparition prématurée d’ostéoporose chez certaines patientes à l’âge adulte. De nos jours, on admet volontiers que la scoliose idiopathique de l’adolescent (SIA) est une maladie d’origine multifactorielle. En effet, des facteurs d’ordre génétique, neurologique, endocrinien, biomécanique, métabolique et autres peuvent intervenir tant dans le déclenchement que dans l’aggravation de cette maladie.
Facteurs étiologiques de la scoliose idiopathique
Plusieurs perspectives divergentes ont été présentées dans l’espoir de définir plus clairement l’étiologie de la SIA et d’expliquer les différentes perturbations associées à la déformation du rachis observée chez les patients SIA [1, 2]. Dans cette synthèse nous proposons un survol des différents facteurs considérés dans l’étiologie de la SIA et nous nous attarderons sur les plus récents concepts moléculaires impliquant un défaut de signalisation de la mélatonine dans l’étiopathogénie de la SIA.
Facteurs génétiques
Des observations de vrais jumeaux atteints de scolioses ont été rapportées à de nombreuses reprises et montrent le développement de scoliose dans 73 % des cas pour les jumeaux identiques et 36 % des cas chez les jumeaux gémellaires [3, 4] confirmant la contribution de facteurs génétiques. Bien que la composante héréditaire puisse être présente dans certaines familles, on note que 85 % des scolioses idiopathiques constituent des cas sporadiques : elles surviennent dans des familles n’ayant aucun antécédent. Néanmoins, plusieurs études ont clairement mis en évidence que la SIA survient avec une fréquence plus élevée dans certaines familles [1]. Les travaux de Wynne-Davies [5] et de DeGeorge [6] ont permis de démontrer une incidence familiale suggérant un mode de transmission dominante et ont d’ailleurs établi une prévalence dans la transmission de la scoliose variant de 17 à 42 % chez les filles et de 7 à 29 % chez les garçons issus d’un parent atteint de SIA sans qu’il y ait une différence significative selon que la mère ou le père soit affecté d’une scoliose [5, 6]. La prédominance de la SIA chez les filles a conduit plusieurs groupes à examiner une possible transmission de la SIA via le chromosome X [7] ; d’autres groupes favorisent plutôt une transmission autosomale [8]. Les plus récentes données génétiques obtenues dans différentes populations ont mis en évidence un nombre croissant de locus de susceptibilité pour la SIA, contribuant ainsi à accroître la difficulté d’identifier un ou plusieurs gènes candidats responsables de cette maladie (Tableau I). Certes ce décompte peut s’expliquer par le nombre limité de familles comprenant plusieurs cas de SIA recrutées à ce jour mais on peut aussi l’attribuer à l’absence de méthodes permettant de mieux classifier les patients scoliotiques. En effet, les analyses génétiques reposent sur le phénotype principal soit la courbure du rachis. Cependant, d’autres paramètres cliniques, biochimiques ou métaboliques (endophénotypes) devraient être pris en compte afin de tirer profit des plus récentes études génétiques et de mieux définir l’étiopathogénie de la SIA.
Facteurs étiologiques de la SIA.
Éléments structuraux du rachis
La scoliose est un des signes cliniques qu’on retrouve dans plusieurs maladies affectant le tissu conjonctif (syndrome de Marfan, hyperlaxité articulaire d’Ehlers-Danlos, homocystinurie et ostéogenèse imparfaite) [9]. Des altérations du collagène seraient aussi reliées à des facteurs environnementaux notamment une insuffisance en cuivre déclenchant une scoliose, car le cuivre intervient dans la régulation de la lysiloxydase, chaînon important du métabolisme du collagène [10]. Dans ce contexte, il n’est donc pas surprenant que les principaux éléments des structures vertébrales, soit le collagène et les protéoglycanes, aient été analysés dans l’espoir d’élucider la cause de la SIA [11, 12]. Ces différents changements de points de vue sont résumés au Tableau 1. En définitive, l’analyse des gènes codant pour ces différentes composantes a permis d’exclure la moindre implication de ces gènes dans l’étiopathogénie de la scoliose [13, 14]. Conséquemment, les modifications observées dans la SIA seraient plutôt des événements secondaires associés à cette maladie : et ils se limiteraient seulement à l’aggravation des déformations.
Facteurs neurologiques et musculaires
Plusieurs preuves expérimentales relèvent des anomalies de fonctionnement des centres du contrôle postural situés dans le cerveau, qui priveraient la colonne vertébrale d’informations nécessaires à sa croissance régulière en hauteur [15]. La mauvaise intégration de ces informations dans le système nerveux central (SNC) ou encore dans le système nerveux périphérique pourrait expliquer la croissance asymétrique du rachis chez les enfants et adolescents atteints de SIA. En effet, la destruction de l’hypothalamus chez le rat par stéréotaxie électrique entraîne la formation d’une scoliose [16]. Une étude réalisée sur 150 sujets scoliotiques a montré que 79 % d’entre eux présentaient des anomalies de fonctionnement de l’équilibre orthostatique portant autant sur la proprioception que sur les réflexes oculomoteurs ; en comparaison, seulement 5 % des sujets témoins présentaient ces anomalies [17]. Le rôle de la musculature spinale dans la pathogénie de la scoliose idiopathique a été également le sujet de nombreuses études, en particulier dans la distribution des différents types de fibres associés à la déformation [18]. Mannion et al. ont montré chez 14 adolescentes scoliotiques qu’il y avait une proportion significativement plus faible de fibres de type I dans les muscles du côté concave de la courbe scoliotique mais pas de différence du côté convexe [19].
Facteurs biochimiques
Les facteurs biochimiques en jeu dans l’étiopathogénie de la scoliose ont surtout été observés dans les plaquettes sanguines qui sont également des cellules présentant un activité contractile [20–22]. Une augmentation du calcium et du phosphore intracellulaire dans les plaquettes de patients SIA, ainsi qu’une diminution de l’activité de plusieurs protéines intracellulaires telles que les protéines contractiles et la myosine adénosine triphosphatase ont été observées [20]. L’augmentation des taux de calmoduline dans les plaquettes de patients SIA serait associée à la SIA et notamment à la progression de la courbure de la scoliose [21]. Plus récemment, les travaux provenant des équipes dirigées par les Drs Enouf à Paris et Moreau à Montréal ont mis en évidence un défaut dans la différenciation et dans la maturation des plaquettes sanguines des sujets atteints de SIA particulièrement en étudiant les profils d’expression de différentes isoformes des ATPases calciques PMCA et SERCA. Cependant, les défauts observés dans les plaquettes représenteraient une indication des changements métaboliques survenant dans d’autres types cellulaires et dans les tissus tels que les muscles et les ostéoblastes plutôt qu’un événement primaire à l’origine de la SIA [20–22].
Facteurs neuroendocriniens
La piste neuroendocrinienne fondée sur l’hypothèse qu’une carence en mélatonine serait à la source de la SIA a suscité beaucoup d’intérêt et de controverses. Cette hypothèse découle du fait que l’ablation de la glande pinéale chez le poulet ou le rat maintenu dans un mode bipède, produit une scoliose similaire en plusieurs points à la maladie humaine [15]. Plus récemment, des résultats identiques ont été obtenus chez des souris bipèdes et quadrupèdes de souche C57B1/6 (souris dont la voie de synthèse de la mélatonine est diminuée à la suite d’une mutation) [23]. En effet, la mélatonine (N-acétyl-5-méthoxytryptamine) est une neurohormone principalement synthétisée par la glande pinéale selon un rythme circadien ; elle est sécrétée dans la circulation où elle agit sur des tissus cibles [24]. La mélatonine est synthétisée également par d’autres cellules notamment les lymphocytes et les plaquettes et régule plusieurs activités physiologiques notamment l’horloge biologique (cycle circadien), le système nerveux et immunitaire de même que le métabolisme osseux. L’injection de doses quotidiennes de mélatonine exogène chez les animaux pinéalectomisés prévient la formation d’une scoliose [23]. La pertinence biologique de la mélatonine dans la SIA est cependant controversée puisque la plupart des études n’ont pu mettre en évidence une diminution significative du taux de mélatonine circulante chez les patients SIA. En outre, ni l’ablation de la glande pinéale chez les animaux supérieurs comme le singe, ni la destruction de la glande pinéale par des traitements de radiothérapie chez les enfants atteints d’un cancer rare de la glande pinéale ne conduisent au développement d’une scoliose : ces fait vont à l’encontre de l’hypothèse d’une carence en mélatonine comme facteur étiologique de la SIA. Voilà pourquoi nous avons été conduits à vérifier si la SIA pouvait compter parmi ses facteurs de déclenchement un défaut de signalisation de la mélatonine dans la cellule.
Signalisation de la mélatonine et SIA
La mélatonine exerce ses effets par le biais de récepteurs spécifiques de haute affinité. Ces récepteurs de la mélatonine sont couplés à des petites protéines appelées protéines G inhibitrices ou Gi (guanine nucleotide-binding protein) et leur activation conduit à une inhibition des adénylates cyclases, enzymes responsables de la synthèse d’AMPc. Par clonage moléculaire, trois sous-types de récepteurs mélatoninergiques couplés aux protéines G ont été identifiés chez les vertébrés [25]. Les propriétés de liaison au ligand, ainsi que les mécanismes de signalisation de ces récepteurs sont remarquablement similaires. Chaque sous-type de récepteur est couplé à une inhibition de l’accumulation d’AMPc. Les gènes des récepteurs MT1 et MT2 sont présents chez les mammifères et de nombreuses données démontrent que MT1 est le récepteur qui régule les fonctions reproductives et circadiennes en réponse à la mélatonine [25]. Le troisième récepteur, MelR1c (deux isoformes α et β) a uniquement été détecté chez le poulet et le Xénope. Un deuxième type de récepteur de la mélatonine, appelé MT3, a d’abord été découvert grâce à ses propriétés pharmacologiques mais il est fort distinct des récepteurs MT1 et MT2 [26]. Récemment, les gènes codant pour les récepteurs MT3 de l’homme et de la souris ont été clonés ; les récepteurs correspondent à une protéine codée par un gène homologue de la quinone réductase 2 (QR2). Le rôle exact de ce gène dans la transmission du signal de la mélatonine reste à déterminer. En dehors des récepteurs membranaires, les récepteurs nucléaires orphelins RZRα et β ont été proposés comme interagissant avec la mélatonine [27]. Nous avons été les premiers à démontrer un dysfonctionnement de la signalisation intracellulaire de la mélatonine dans les tissus musculo-squelettiques d’une cohorte de patients atteints de SIA [28] que nous avons comparée à une série de sujets témoins atteints ou non d’un autre type de scoliose cliniquement reconnu et décrit. Les tests que nous avons effectués ont permis d’observer trois types de réponses en présence de concentrations croissantes de mélatonine suggérant ainsi la participation possible de gènes distincts dans ce défaut de signalisation chez certains patients SIA (Figure 1A). De plus, la faible capacité à inhiber l’activité des adénylates cyclases stimulées par la forskoline a également été observée avec un analogue non hydrolysable du GTP, le Gpp(NH)p, permettant de penser qu’un tel dysfonctionnement de la signalisation de la mélatonine dans la SIA pourrait être induit par une hypo-fonctionnalité sélective des protéines Gi, fait d’ailleurs accrédité par une augmentation de la phosphorylation des protéines Gi couplées aux récepteurs de la mélatonine. En effet, les modifications post-traductionnelles des protéines Gi impliquant la phosphorylation de résidus sérine à leur extrémité amino-terminale sont bien connues pour bloquer la formation d’hétérotrimères fonctionnels avec les sous-unités Gβ et Gγ, empêchant ainsi l’inhibition de l’activité des adénylates cyclases en présence d’un ligand spécifique [29]. Cette phosphorylation anormale nichée dans les ostéoblastes et dans plusieurs autres types cellulaires testés témoignant d’un défaut systémique (Figure 1B). Fait intéressant, les œstrogènes inhibent fortement l’expression et la synthèse de la sous-unité α des protéines G (Gi1-3 et Gs) dans des cultures d’ostéoblastes [30], suggérant ainsi que la signalisation de la mélatonine pourrait être modulée par les œstrogènes. On peut remarquer que la capacité des œstrogènes à moduler les niveaux des protéines G (Gs et Gi) pourrait expliquer l’incidence des manifestations cliniques de la SIA à l’adolescence et le fait que les filles soient plus sévèrement atteintes que les garçons. Dans ce sens, la mise en évidence d’un défaut de signalisation de la mélatonine vient non seulement actualiser le rôle de cette hormone dans la pathogénie de la scoliose mais, de plus, constitue un concept unificateur pouvant expliquer la majorité des affections associées à la scoliose (Figure 2). De plus, ce nouveau concept moléculaire a permis de développer un premier test de dépistage fonctionnel de la SIA actuellement en cours de validation clinique.
 |
Figure 1. A. Évaluation de l’effet inhibiteur de la mélatonine sur l’accumulation intracellulaire d’AMPc dans les ostéoblastes humains. Effet de doses croissantes de mélatonine (10–11 à 10–5 M) sur les niveaux d’AMPc intracellulaire induit par la stimulation de l’activité adénylate cyclase par la forskoline (10–5 M) dans les ostéoblastes dérivés de patients scoliotiques. Les courbes d’inhibition ont permis de classifier les patients SIA en trois groupes fonctionnels. Le groupe 1 (
|



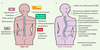 |
Figure 2. Systèmes ciblés par l’action de la mélatonine et symptômes observés chez les patients scoliotiques. Représentation schématique des effets physiologiques de la mélatonine chez les sujets normaux et leurs liens avec les pathologies observées chez les patients SIA. A. Tissus ciblés par l’action de la mélatonine. Une défaillance de la signalisation de cette hormone dans ces systèmes pourrait expliquer la majorité des anomalies et des symptômes observés chez les patients SIA (B), ce qui confère à ce modèle la portée d’un concept unificateur pour expliquer l’étiologie de la SIA. |
Développement d’un test fonctionnel pour le dépistage précoce de la SIA
Comme il n’existe pour le moment aucun test ni une quelconque façon de prévoir l’apparition d’une scoliose, celle-ci n’est diagnostiquée que lorsque la déviation de la colonne commence à être apparente. Ainsi c’est toute la manière dont on traite les patients scoliotiques depuis des décennies qui sera bouleversée, voire transformée, par la mise au point d’un test de dépistage précoce. Le test cible deux populations distinctes. Premièrement, les enfants dits à risque, c’est-à-dire ceux provenant de familles présentant plusieurs cas de scoliose ou encore ceux dont l’un des deux parents est scoliotique [5, 6]. Deuxièmement les enfants et les adolescents en début de maladie (c’est-à-dire, victimes d’une déformation du rachis apparente comprise entre 10° et 30°) au sein desquels il sera possible d’identifier ceux qui présentent un risque de progression rapide. Le test est relativement simple puisque à partir d’un échantillon sanguin, les lymphocytes possédant des récepteurs de la mélatonine, sont isolés et utilisés pour déterminer la présence d’un défaut dans la transmission de la mélatonine. Ce défaut est déterminé par un test biochimique mesurant la capacité de la mélatonine à inhiber la synthèse d’AMPc, molécule chimique produite par la cellule et agissant comme messager intracellulaire secondaire (Figure 1). La production d’AMPc est artificiellement stimulée par un traitement en présence de forskoline, molécule qui stimule l’activité des adénylate cyclases, enzymes membranaires chargées de la synthèse d’AMPc. En présence de concentrations croissantes de mélatonine, on observe une réduction rapide de cette synthèse dans les lymphocytes des sujets sains ; en revanche, dans les lymphocytes des patients SIA, on observe une absence d’inhibition. Nous avons identifié trois types de réponses anormales chez les sujets SIA ce qui nous permet de faire une classification fonctionnelle des patients. En outre, ce test fonctionnel permet le dépistage précoce de la SIA sans avoir a priori une connaissance des mutations ou des gènes en cause dans la SIA puisqu’il cible une voie de signalisation ce qui couvre un ensemble plus général.
Remerciements
Nous tenons à remercier sincèrement les patients et leur famille qui ont participé à cette étude, de même que les cliniciens qui nous ont référé ces patients. Ce projet de recherche est subventionné par la Fondation Yves Cotrel de l’Institut de France (Drs Moreau et Labelle) et la validation des tests cliniques par la société Paradigm Spine de New York (Dr Moreau).
Références
- Cowell HR, Hall JN, MacEwen GD. Genetic aspects of idiopathic scoliosis. A Nicholas Andry Award essay, 1970. Clin Orthop Relat Res 1972; 86 : 121–31. [Google Scholar]
- Lowe TG, Edgar M, Margulies JY, et al. Etiology of idiopathic scoliosis : current trends in research. J Bone Joint Surg Am 2000; 82-A : 1157–68. [Google Scholar]
- Carr AJ. Adolescent idiopathic scoliosis in identical twins. J Bone Joint Surg Br 1990; 72 : 1077. [Google Scholar]
- Burwell RG. Aetiology of idiopathic scoliosis : current concepts. Pediatr Rehabil 2003; 6 : 137–70. [Google Scholar]
- Wynne-Davies R. Familial (idiopathic) scoliosis. A family survey. J Bone Joint Surg Br 1968; 50 : 24–30. [Google Scholar]
- De George FV, Fisher RL. Idiopathic scoliosis : genetic and environmental aspects. J Med Genet 1967; 4 : 251–7. [Google Scholar]
- Justice CM, Miller NH, Marosy B, et al. Familial idiopathic scoliosis : evidence of an X-linked susceptibility locus. Spine 2003; 28 : 589–94. [Google Scholar]
- Salehi LB, Mangino M, De SS, et al. Assignment of a locus for autosomal dominant idiopathic scoliosis (IS) to human chromosome 17p11. Hum Genet 2002; 111 : 401–4. [Google Scholar]
- Pyeritz RE, McKusick VA. The Marfan syndrome : diagnosis and management. N Engl J Med 1979; 300 : 772–7. [Google Scholar]
- Dastych M, Cienciala J. Idiopathic scoliosis and concentrations of zinc, copper, and selenium in blood plasma. Biol Trace Elem Res 2002; 89 : 105–10. [Google Scholar]
- Hadley-Miller N, Mims B, Milewicz DM. The potential role of the elastic fiber system in adolescent idiopathic scoliosis. J Bone Joint Surg Am 1994; 76 : 1193–206. [Google Scholar]
- Akhtar S, Davies JR, Caterson B. Ultrastructural localization and distribution of proteoglycan in normal and scoliotic lumbar disc. Spine 2005; 30 : 1303–9. [Google Scholar]
- Miller NH, Mims B, Child A, et al. Genetic analysis of structural elastic fiber and collagen genes in familial adolescent idiopathic scoliosis. J Orthop Res 1996; 14 : 994–9. [Google Scholar]
- Carr AJ, Ogilvie DJ, Wordsworth BP, et al. Segregation of structural collagen genes in adolescent idiopathic scoliosis. Clin Orthop Relat res 1992; 274 : 305–10. [Google Scholar]
- Dubousset J, Machida M. Possible role of the pineal gland in the pathogenesis of idiopathic scoliosis. Experimental and clinical studies. Bull Acad Natl Med 2001; 185 : 593–602. [Google Scholar]
- Yamada K, Ikata T, Yamamoto H, et al. Equilibrium function in scoliosis and active corrective plaster jacket for the treatment. Tokushima J Exp Med 1969; 16 : 1–7. [Google Scholar]
- Yamada K, Yamamoto H, Nakagawa Y, et al. Etiology of idiopathic scoliosis. Clin Orthop Relat Res 1984; 184 : 50–7. [Google Scholar]
- Porter RW. The pathogenesis of idiopathic scoliosis : uncoupled neuro-osseous growth ? Eur Spine J 2001; 10 : 473–81. [Google Scholar]
- Mannion AF, Meier M, Grob D, et al. Paraspinal muscle fibre type alterations associated with scoliosis : an old problem revisited with new evidence. Eur Spine J 1998; 7 : 289–93. [Google Scholar]
- Yarom R, Meyer S, More R, et al. Metal impregnation abnormalities in platelets of patients with idiopathic scoliosis. Haemostasis 1982; 12 : 282–8. [Google Scholar]
- Kindsfater K, Lowe T, Lawellin D, et al. Levels of platelet calmodulin for the prediction of progression and severity of adolescent idiopathic scoliosis. J Bone Joint Surg Am 1994; 76 : 1186–92. [Google Scholar]
- Machida M, Dubousset J, Imamura Y, et al. Melatonin. A possible role in pathogenesis of adolescent idiopathic scoliosis. Spine 1996; 21 : 1147–52. [Google Scholar]
- Machida M, Dubousset J, Yamada T, et al. Experimental scoliosis in melatonin-deficient C57BL/6J mice without pinealectomy. J Pineal Res 2006; 41 : 1–7. [Google Scholar]
- Macchi MM, Bruce JN. Human pineal physiology and functional significance of melatonin. Front Neuroendocrinol 2004; 25 : 177–95. [Google Scholar]
- Witt-Enderby PA, Bennett J, Jarzynka MJ, et al. Melatonin receptors and their regulation : biochemical and structural mechanisms. Life Sci 2003; 72 : 2183–98. [Google Scholar]
- Nosjean O, Ferro M, Coge F, et al. Identification of the melatonin-binding site MT3 as the quinone reductase 2. J Biol Chem 2000; 275 : 31311–7. [Google Scholar]
- Smirnov AN. Nuclear melatonin receptors. Biochemistry (Mosc) 2001; 66 : 19–26. [Google Scholar]
- Moreau A, Wang DS, Forget S, et al. Melatonin signaling dysfunction in adolescent idiopathic scoliosis. Spine 2004 29 : 1772–81. [Google Scholar]
- Strassheim D, Malbon CC. Phosphorylation of Gi alpha 2 attenuates inhibitory adenylyl cyclase in neuroblastoma/glioma hybrid (NG-108-15) cells. J Biol Chem 1994; 269 : 14307–13. [Google Scholar]
- Papaioannou S, Tumber AM, Meikle MC, et al. G-protein signalling pathways and oestrogen : a role of balanced maintenance in osteoblasts. Biochim Biophys Acta 1999; 1449 : 284–92. [Google Scholar]
- Oegema TR, Jr, Bradford DS, Cooper KM, et al. Comparison of the biochemistry of proteoglycans isolated from normal, idiopathic scoliotic and cerebral palsy spines. Spine 1983; 8 : 378–84. [Google Scholar]
- Duance VC, Crean JK, Sims TJ, et al. Changes in collagen cross-linking in degenerative disc disease and scoliosis. Spine 1998; 23 : 2545–51. [Google Scholar]
- Yu J, Fairbank JC, Roberts S, et al. The elastic fiber network of the anulus fibrosus of the normal and scoliotic human intervertebral disc. Spine 2005; 30 : 1815–20. [Google Scholar]
- Bagnall KM, Beuerlein M, Johnson P, et al. Pineal transplantation after pinealectomy in young chickens has no effect on the development of scoliosis. Spine 2001; 26 : 1022–7. [Google Scholar]
- Brodner W, Krepler P, Nicolakis M, et al. Melatonin and adolescent idiopathic scoliosis. J Bone Joint Surg Br 2000; 82 : 399–403. [Google Scholar]
- Morcuende JA, Minhas R, Dolan L, et al. Allelic variants of human melatonin 1A receptor in patients with familial adolescent idiopathic scoliosis. Spine 2003; 28 : 2025–8. [Google Scholar]
- Wise CA, Barnes R, Gillum J, et al. Localization of susceptibility to familial idiopathic scoliosis. Spine 2000; 25 : 2372–80. [Google Scholar]
- Chan V, Fong GC, Luk KD, et al. A genetic locus for adolescent idiopathic scoliosis linked to chromosome 19p13.3. Am J Hum Genet 2002; 71 : 401–6. [Google Scholar]
- Alden KJ, Marosy B, Nzegwu N, et al. Idiopathic scoliosis : identification of candidate regions on chromosome 19p13. Spine 2006; 31 : 1815–9. [Google Scholar]
- Shapirov RN, Zaidman AM, Zorkol’tseva IV, et al. Polymorphism of aggrecan gene in families with idiopathic scoliosis. Mol Biol (Mosk) 2006; 40 : 554–7. [Google Scholar]
- Bashiardes S, Veile R, Allen M, et al. SNTG1, the gene encoding gamma1-syntrophin : a candidate gene for idiopathic scoliosis. Hum Genet 2004; 115 : 81–9. [Google Scholar]
Liste des tableaux
Liste des figures
|
Figure 1. A. Évaluation de l’effet inhibiteur de la mélatonine sur l’accumulation intracellulaire d’AMPc dans les ostéoblastes humains. Effet de doses croissantes de mélatonine (10–11 à 10–5 M) sur les niveaux d’AMPc intracellulaire induit par la stimulation de l’activité adénylate cyclase par la forskoline (10–5 M) dans les ostéoblastes dérivés de patients scoliotiques. Les courbes d’inhibition ont permis de classifier les patients SIA en trois groupes fonctionnels. Le groupe 1 (
|
| Dans le texte | |
|
Figure 2. Systèmes ciblés par l’action de la mélatonine et symptômes observés chez les patients scoliotiques. Représentation schématique des effets physiologiques de la mélatonine chez les sujets normaux et leurs liens avec les pathologies observées chez les patients SIA. A. Tissus ciblés par l’action de la mélatonine. Une défaillance de la signalisation de cette hormone dans ces systèmes pourrait expliquer la majorité des anomalies et des symptômes observés chez les patients SIA (B), ce qui confère à ce modèle la portée d’un concept unificateur pour expliquer l’étiologie de la SIA. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.