")
")
| Issue |
Med Sci (Paris)
Volume 23, Number 6-7, Juin-Juillet 2007
|
|
|---|---|---|
| Page(s) | 581 - 583 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/20072367581 | |
| Published online | 15 juin 2007 | |
P53 sous le soleil
Suntanning and p53
CNRS, Équipe Génétique et physiopathologie des cancers, épidermiques, FRE Génomes et Cancer, Institut Gustave-Roussy, PR2, 39, rue Camille-Desmoulins, 94805 Villejuif Cedex France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
P53 et stress génotoxique
Le gène P53 est un gène suppresseur de tumeur codant pour un facteur de transcription subissant de nombreuses modifications post-traductionnelles et impliqué dans le contrôle du cycle cellulaire et de l’apoptose (mort cellulaire programmée). P53 est muté dans plus de 50 % des tumeurs humaines, notamment dans les tumeurs de la peau induites par les rayonnements ultraviolets (UV) contenus dans la lumière solaire. Les travaux récemment publiés par l’équipe de David Fisher à Harvard ont attribué à la protéine P53 une nouvelle mission de surveillance et de contrôle de la pigmentation cutanée normale et pathologique [1].
Identifiée il y bientôt 30 ans [2–4], P53 est probablement l’une des protéines de régulation les plus étudiées (plus de 40 000 études font référence à P53) Il semble aujourd’hui que P53 assure son rôle de « gardien du génome » [5] essentiellement au travers d’une multiplicité de régulations de la transcription visant à protéger la cellule contre les agressions extérieures [6].
Les UV constituent le stress génotoxique majeur des cellules de la peau. Les UV introduisent des lésions dans l’ADN (dimères de pyrimidines) dont la réplication peut être mutagène et à l’origine du développement tumoral [7]. Le destin cellulaire peut être déterminé par la quantité des lésions accumulées dans l’ADN. Ainsi, un nombre raisonnable - c’est-à-dire qui ne sature pas les systèmes de réparation de l’ADN - de lésions dans l’ADN provoque la stabilisation de P53, l’arrêt du cycle cellulaire (G1/S), et régule l’expression de certains gènes de réparation de l’ADN, notamment XPC (xeroderma pigmentosum, complementation group c) [8]. Ce dernier est indispensable à la reconnaissance des dimères de pyrimidine induits par les UV dans l’ADN [9]. Si la quantité des lésions dans l’ADN est telle que les systèmes de réparation ne peuvent y faire face, la cellule lésée est orientée vers l’apoptose. En 1997, les travaux du groupe de Bert Vogelstein à Baltimore ont indiqué que certains des gènes précocement induits au cours de l’apoptose dépendante de P53 codent pour des protéines génératrices d’espèces réactives de l’oxygène (ROS) pour promouvoir l’élimination de la cellule lésée [10]. Sauveur ou tueur, P53 protège notre organisme soit en contribuant à l’élimination des lésions dans l’ADN, soit en participant à l’élimination des cellules potentiellement tumorales. Le rôle de P53 n’est toutefois pas limité aux situations de stress génotoxique exogènes. Des travaux plus récents ont ainsi rapporté que l’expression de P53 à bas niveau est nécessaire à l’élimination des ROS produites en permanence par le métabolisme cellulaire et qui provoquent, elles aussi, des dommages dans l’ADN (bases oxydées, cassures simple brin de l’ADN) [11]. Ces travaux montrent que l’inactivation expérimentale de P53 favorise l’accumulation de ROS. Les mutations de P53 qui altèrent sa capacité à réguler l’expression des gènes pourraient contribuer à l’accumulation de ROS et favoriser l’instabilité génétique des tumeurs. L’ensemble de ces observations indique que la surveillance du génome par P53 est indispensable, diversifiée et adaptée en fonction du type et de la quantité des lésions génotoxiques.
P53 et pigmentation
La pigmentation cutanée joue également un rôle protecteur contre les tumeurs de la peau, cancers de plus forte incidence chez l’homme (carcinomes baso- et spinocellulaires). Ces cancers se développent à partir des kératinocytes qui sont les cellules majoritaires de l’épiderme, le tissu superficiel de la peau. L’exposition aux UV de la lumière solaire stimule la production de mélanines par les mélanocytes. Ces cellules spécialisées (dérivées des crêtes neurales) sont situées dans la couche basale de l’épiderme qui abrite aussi les cellules souches épidermiques (Figure 1A). Les mélanines sont transmises aux kératinocytes sous forme de petits granules appelés mélanosomes (Figure 1B). La pigmentation assure ainsi une protection naturelle du génome kératinocytaire en absorbant les ultraviolets [12]. L’importance de la protection assurée par les mélanines est illustrée par des taux de cancers cutanés significativement plus élevés chez les personnes à peau claire que chez les personnes à peau plus sombre. Dans les populations australiennes d’ascendance celte, de pigmentation claire et qui souvent brûlent plus qu’elles ne bronzent, les cancers de la peau induits par les UV constituent un réel problème de santé publique.
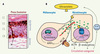 |
Figure 1. A. Coupe de peau humaine à forte pigmentation. L’épiderme est en majeure partie composé de kératinocytes. M indique un mélanocyte situé dans la couche basale de l’épiderme (CB). CS : couches spineuses ; CG : couches granuleuses ; CC : couches cornées. Le derme sous-jacent, et l’épiderme superficiel sont indiqués. B. L’exposition aux ultraviolets conduit à la stabilisation de P53 (tétramère) qui va alors activer la transcription du gène POMC. Le peptide précurseur POMC subit une maturation pour produire les peptides α-MSH, ACTH, et β-endorphine. L’α-MSH sécrétée en plus grande partie par les kératinocytes se fixe au récepteur MC1R, ce qui conduit à l’augmentation du niveau d’AMPc et à l’activation du facteur de transcription MITF (microphtalmia associated transcription factor) qui va activer la transcription du gène de la tyrosinase, ce qui conduit à la production de mélanines (granules de deux types). Ces granules sont distribués aux kératinocytes par l’intermédiaire des dendrites mélanocytaires. La sécrétion de la β-endorphine dans la circulation sanguine pourrait contribuer à la recherche de bien-être procuré par l’exposition solaire. |
La pigmentation cutanée dépend des interactions cellulaires entre kératinocytes et mélanocytes. L’exposition aux UV stimule la transcription du gène appelé POMC (pro-opio-melano-cortin complex) (Figure 1B) codant pour l’hormone α-MSH (α-melanocyte stimulating hormone), l’ACTH (adreno corticotrophic hormone) ainsi que la β-endorphine. La production d’α-MSH est spécifique des tissus cutanés et est observée dans les kératinocytes et dans les mélanocytes, notamment après exposition UV [1]. Une fois produite et sécrétée, l’α-MSH se fixe au récepteur membranaire MC1R (mélanocortine récepteur 1) présent à la surface des mélanocytes. MC1R active la protéine kinase A, conduisant à la production accrue d’AMPc (adénosyl monophosphate cyclique) puis à l’activation de facteurs de transcription spécifiques contrôlant la synthèse des mélanines. Ces dernières sont ensuite distribuées aux kératinocytes qui se trouvent ainsi protégés. Toutefois, la mécanistique de cette régulation restait assez mal caractérisée jusqu’à la publication de D. Fisher. Les observations de D. Fisher partent d’une analyse in silico des régions de l’ADN génomique impliquées dans la régulation de la transcription du gène POMC. Ces analyses ont révélé une séquence d’ADN régulatrice fonctionnelle, cible de la protéine suppresseur de tumeur P53. De plus, les travaux montrent que la transcription de POMC (et donc la production d’α-MSH) est dépendante de la stabilisation de P53 après un stress génotoxique et qu’elle est quantitativement beaucoup plus importante dans les kératinocytes que dans les mélanocytes. L’invalidation du gène P53 chez la souris (souris p53−/−) abolit l’augmentation du transcrit POMC après UV et, de fait, le bronzage (limité aux oreilles chez la souris). Ces données suggèrent que la régulation de la pigmentation cutanée après UV fait l’objet d’interactions cellulaires étroites entre les kératinocytes et les mélanocytes. Cette hypothèse est soutenue par le fait que, dans l’épiderme humain, un mélanocyte interagit avec environ 35 kératinocytes pour former une « unité de mélanogenèse ». La capacité de pigmentation de ces unités apparaît donc étroitement liée à la réponse des kératinocytes aux UV. Dans les mélanocytes, il a été montré que la transcription du gène codant pour l’enzyme clé de la mélanogenèse (la tyrosinase) est aussi stimulée par l’exposition aux UV [13]. Dans ce cas, la réponse transcriptionnelle est plus lente que pour POMC, et résulte probablement d’un mécanisme de régulation indirect.
Les aspects physiopathologiques de la régulation de la production d’α-MSH par P53 ont été abordés de façon très intéressante par l’équipe de D. Fisher au travers de l’analyse du statut de P53 dans des carcinomes basocellulaires pigmentés ou non pigmentés. Toutes les tumeurs non pigmentées étudiées présentaient au moins une mutation de P53 alors que les tumeurs pigmentées étaient systématiquement normales pour P53. Ces observations suggèrent que la stabilisation constitutive de P53 sauvage, déjà décrite dans certaines tumeurs humaines [14], conduit, via la sécrétion de α-MSH, au recrutement des mélanocytes environnants. Ce mécanisme pourrait limiter l’instabilité génétique décrite dans les tumeurs en l’absence d’une protéine P53 fonctionnelle.
Ces travaux suggèrent aussi que la capacité de P53 à activer la transcription du gène POMC est liée au potentiel de pigmentation individuel. Ainsi, le variant P53 72Pro qui est un activateur de la transcription plus puissant que la forme plus répandue P53 72Arg pourrait être plus favorable à la pigmentation UV-induite. Toutefois, il ne semble pas exister de corrélation entre le polymorphisme P53 72Pro et l’incidence des cancers d’origine kératinocytaire [15].
Conclusions
La découverte du rôle direct de la stabilisation de P53 dans la pigmentation pourrait susciter des recherches pharmacologiques visant à joindre l’utile à l’agréable : protection du génome et bronzage. L’étude publiée montre notamment que P53 stabilisée stimule l’expression de l’ensemble du complexe POMC, ce qui aboutit à la sécrétion du peptide opiacé β-endorphine et peut-être au bien-être naturel qui accompagne l’exposition au soleil (sun seeking behavior). Manipuler l’expression de POMC est donc une hypothèse séduisante, mais elle est, bien entendu, à envisager avec précaution.
Remerciements
Je remercie le Dr Françoise Bernerd et Emilie Warrick pour leurs discussions et leur lecture critique.
Références
- Cui R, Widlund HR, Feige E, et al. Central role of p53 in the suntan response and pathologic hyperpigmentation. Cell 2007; 128 : 853–64. [Google Scholar]
- Linzer DI, Levine AJ. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 1979; 17 : 43–52. [Google Scholar]
- Lane DP, Crawford LV. T antigen is bound to a host protein in SV40-transformed cells. Nature 1979; 278 : 261–3. [Google Scholar]
- Kress M, May E, Cassingena R, May P. Simian virus 40-transformed cells express new species of proteins precipitable by anti-simian virus 40 tumor serum. J Virol 1979; 31 : 472–83. [Google Scholar]
- Lane DP. Cancer. p53, guardian of the genome. Nature 1992; 358 : 15–6. [Google Scholar]
- Toledo F, Bluteau O, Simeonova I. Réactivation de p53 dans les tumeurs : une stratégie antitumorale prometteuse. Med Sci (Paris) 2007; 23 : 565–7. [Google Scholar]
- Magnaldo T. La «guerre» du NER (nucleotide excision repair). Med Sci (Paris) 2004; 20 : 268–70. [Google Scholar]
- Adimoolam S, Ford JM. p53 and DNA damage-inducible expression of the xeroderma pigmentosum group C gene. Proc Natl Acad Sci USA 2002; 99 : 12985–90. [Google Scholar]
- Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature 2001; 411 : 366–74. [Google Scholar]
- Polyak K, Xia Y, Zweier JL, et al. A model for p53-induced apoptosis. Nature 1997; 389 : 300–5. [Google Scholar]
- Sablina AA, Budanov AV, Ilyinskaya GV, et al. The antioxidant function of the p53 tumor suppressor. Nat Med 2005; 11 : 1306–13. [Google Scholar]
- Miyamura Y, Coelho SG, Wolber R, et al. Regulation of human skin pigmentation and responses to ultraviolet radiation. Pigment Cell Res 2007; 20 : 2–13. [Google Scholar]
- Khlgatian MK, Hadshiew IM, Asawanonda P, et al. Tyrosinase gene expression is regulated by p53. Jof Inv Dermatol 2002; 118 : 126–32. [Google Scholar]
- Bartkova J, Horejsi Z, Koed K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005; 434 : 864–70. [Google Scholar]
- Bendesky A, Rosales A, Salazar AM, et al. p53 codon 72 polymorphism, DNA damage and repair, and risk of non-melanoma skin cancer. Mutat Res 2007; 619 : 38–44. [Google Scholar]
© 2007 médecine/sciences - Inserm / SRMS
Liste des figures
|
Figure 1. A. Coupe de peau humaine à forte pigmentation. L’épiderme est en majeure partie composé de kératinocytes. M indique un mélanocyte situé dans la couche basale de l’épiderme (CB). CS : couches spineuses ; CG : couches granuleuses ; CC : couches cornées. Le derme sous-jacent, et l’épiderme superficiel sont indiqués. B. L’exposition aux ultraviolets conduit à la stabilisation de P53 (tétramère) qui va alors activer la transcription du gène POMC. Le peptide précurseur POMC subit une maturation pour produire les peptides α-MSH, ACTH, et β-endorphine. L’α-MSH sécrétée en plus grande partie par les kératinocytes se fixe au récepteur MC1R, ce qui conduit à l’augmentation du niveau d’AMPc et à l’activation du facteur de transcription MITF (microphtalmia associated transcription factor) qui va activer la transcription du gène de la tyrosinase, ce qui conduit à la production de mélanines (granules de deux types). Ces granules sont distribués aux kératinocytes par l’intermédiaire des dendrites mélanocytaires. La sécrétion de la β-endorphine dans la circulation sanguine pourrait contribuer à la recherche de bien-être procuré par l’exposition solaire. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.