")
")
| Issue |
Med Sci (Paris)
Volume 23, Number 3, Mars 2007
|
|
|---|---|---|
| Page(s) | 297 - 302 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/2007233297 | |
| Published online | 15 March 2007 | |
Génétique moléculaire de MTHFR
Les polymorphismes ne sont pas tous bénins
Molecular genetics of MTHFR: polymorphisms are not all benign
Départements de Génétique Humaine et de Pédiatrie,Université McGill,CUSM-Hôpital de Montréal pour enfants,4060, Sainte-Catherine Ouest,2e étage, Montréal (Québec),H3Z 2Z3 Canada
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
Les polymorphismes génétiques résultent de mutations communes habituellement ignorées par beaucoup de chercheurs car elles sont souvent bénignes.Toutefois, certains polymorphismes ne sont pas sans conséquence sur la santé, comme l’illustre bien le variant 677C → T du gène de la 5,10-méthylène tétrahydrofolate réductase (MTHFR). La MTHFR est une importante enzyme régulatrice du métabolismedes folates (dérivés de l’acide folique/vitamine B9) et de l’homocystéine. Au cours de la récente décennie, de nombreux travaux ont considérablementamélioré notre compréhension des anomalies de MTHFR qui causent, selon la nature de la variation, l’homocystinurie classique ou une légère hyperhomocystéinémie. Le clonage de MTHFR par notre groupe a d’abord permis l’identification de mutationsgraves qui sont particulières aux familles de patients homocystinuriques souffrant d’une hyperhomocystéinémieprononcée. Par la suite, le polymorphisme677C → T a été caractérisé comme un variant produisant une enzyme thermolabile dont l’activité est restreinte. Ce polymorphisme est la cause génétique la plus fréquente d’hyperhomocystéinémie et risque de constituer l’un des facteurs qui participent à certaines affections multifactorielles, comme les maladies cardiovasculaires. Cet article a pour objectif de résumer les connaissances portant sur le gène MTHFR et ses mutations, ainsique sur les découvertes accomplies en utilisant des souris déficientes en MTHFR.
Abstract
Methylenetetrahydrofolate reductase (MTHFR) is a key regulatory enzyme in folate and homocysteine metabolism.Research performed during the past decade has clarified our understanding of MTHFR deficiencies that cause homocystinuria or mild hyperhomocysteinemia. Our cloning of the MTHFR coding sequence was initially followed by the identification of the first deleterious mutation sin MTHFR, in patients with homocystinuria and marked hyperhomocysteinemia. Shortly thereafter, we identified the 677C→Tvariant and showed that it encoded a thermolabile enzyme with reduced activity. Currently, a total of 41 rare but deleterious mutations in MTHFR, as well as about 60 polymorphisms have been reported. The 677C→T (Ala222Val) variant has been particularly noteworthy since it has become recognized as the most common genetic cause of hyperhomocysteinemia. The disruption of homocysteine metabolism by this polymorphism influences risk for several complex disorders, including cardiovascular disease, neural tube defects and some cancers. We describe here the complex structure of the MTHFR gene,summarize the current state of knowledge on rare and common mutations in MTHFR and discuss some relevant findings in a mouse model for MTHFR deficiency.
© 2007 médecine/sciences - Inserm / SRMS
La 5,10-méthylène tétrahydrofolate réductase (MTHFR ; EC 1.5.1.20) joue un rôle crucial dans le métabolisme des folates et de l’homocystéine (Figure 1, chapitre 1 de [1]). Le rôle de la MTHFR dans certaines maladies a d’abord été rapporté par Mudd et al. [2] qui avaient identifié une altération fonctionnelle majeure de cette enzyme chez un patient présentant une homocystinurie. Ce type d’altération génétique est rare (environ 85 cas répertoriés mondialement ; chapitre 4 de [1]). En 1988, une variante commune et thermolabile de la protéine MTHFR fut repérée [3]. La caractérisation de l’ADNc de MTHFR [4–6] nous a permis de répertorier plusieurs mutations de MTHFR chez des patients homocystinuriques, ainsi qu’un variant commun, 677C → T (A222V), qui correspond à la forme thermolabile de l’enzyme [5]. Ce polymorphisme génétique (une mutation communément observée dans la population normale) est la cause génétique la plus fréquente d’hyperhomocystéinémie. Il a été beaucoup étudié et cité (Figure 2). Nous allons présenter une vue d’ensemble des mutations de MTHFR, en insistant sur le polymorphisme 677C → T. Nous allons aussi brièvement décrire le phénotype de la souris dont le gène Mthfr a été inactivé. Il s’agit du modèle animal qui sert à étudier les défauts majeurs ou modérés de la MTHFR.
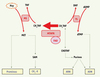 |
Figure 1. Répercussions métaboliques de l’activité de la MTHFR. La 5,10-méthylène tétrahydrofolate réductase (MTHFR) catalyse la réduction irréversible du 5,10-methylène tétrahydrofolate (CH2THF) en 5-méthyltétrahydrofolate (CH3THF). L’activité de la MTHFR affecte ainsi la disponibilité du CH2THF, ce qui influence la synthèse de l’ARN et de l’ADN. Le CH3THF est requis pour la reméthylation de l’homocystéine (Hcy) en méthionine (MET), qui intervient elle-même dans la synthèse protéique et la méthylation de l’ADN et d’autres composés (CH3-X). Le FAD est le cofacteur de la MTHFR. MS, méthionine synthase ; TS, thymidylate synthase ; THF, tétrahydrofolate ; DHF, dihydrofolate ; CHOTHF, 10-formyltétrahydrofolate ; SAM, S-adénosyl méthionine. |
 |
Figure 2. Fréquence annuelle des publications traitant de la MTHFR entre 1988 et 2005. Le nombre d’articles scientifiques discutant de la MTHFR (cercles bleus) a été estimé en interrogeant la plate-forme Ovid MEDLINE à l’aide des termes « (MTHFR OR methylenetetrahydrofolate reductase) ». Les articles énonçant plus spécifiquement des mutations de MTHFR ont été identifiés en utilisant les mots-clés « (MTHFR OR methylenetetrahydrofolate reductase) AND (polymorphism OR 677 OR C677T OR variant OR mutation OR genotype OR risk factor OR thermolabile) » (carrés rouges). L’examen sommaire d’un échantillon représentatif de ces publications montre que la quasi-totalité d’entre elles mentionnent le polymorphisme 677C → T. |
ADNc et structure génomique de MTHFR
Clonage de la séquence codante de MTHFR
Un ADNc de 90 pb fut amplifié par RT-PCR à partir d’ARN de porc [4]. Cet amplicon a été utilisé pour sonder une banque d’ADNc humains, ce qui a permis d’obtenir un ADNc de 1,3 kpb. Une autre banque d’ADNc a été criblée pour découvrir la portion carboxy-terminale de la séquence codante [5]. L’ADNc ainsi déduit représentait 2,2 kpb et comprenait 11 exons (chapitre 1 de [1]).
La MTHFR est un dimère de 150 kDa comprenant deux isoformes de tailles variables : 77 kDa et 70 kDa. L’expression de l’ADNc humain de 2,2 kpb donne une protéine de 70 kDa [5]. Le site de démarrage de la traduction de l’isoforme de 77 kDa permet l’ajout de codons additionnels en amont de la séquence de l’isoforme de 70 kDa [6].
Cartographie
Le gène MTHFR humain a été localisé à la position 1p36.3 [4]. Mthfr se situe dans la portion distale du chromosome 4 de la souris (chapitre 1 de [1]). Le génome de souris contient un pseudogène, Mthfr-ps. Le génome humain ne possède pas de pseudogène de MTHFR (chapitre 1 de [1]).
Produits de transcription et promoteurs
Plusieurs sites d’amorce de la transcription, d’épissage alternatif et de polyadénylation ont été observés pour MTHFR et Mthfr [6]. Les sites de début de la transcription sont localisés dans deux régions et deux promoteurs ont été caractérisés [7]. Leur efficacité relative dépend du type cellulaire [7].
Mutations
Mutations sévères
Les symptômes des patients homocystinuriques (OMIM 236250) se manifestent notamment par des retards de développement, ainsi que par diverses affections neurologiques et vasculaires : neuropathie périphérique, convulsions, thromboses et lésions vasculaires (chapitre 4 de [1]). L’activité enzymatique résiduelle des fibroblastes des patients et l’âge d’apparition des symptômes sont corrélés.
La Figure 3 présente 41 mutations (numérotation en noir) qui sont propres aux familles des patients homocystinuriques répertoriés. Plusieurs mutations ont été exprimées en utilisant des systèmes bactériens ou des systèmes de levures, afin de vérifier leur effet sur l’activité enzymatique (chapitre 1 de [1]). Comme chacune de ces mutations graves n’est généralement observée que dans une ou deux familles, il peut être avantageux de procéder à un diagnostic moléculaire prénatal par étude de liaisons génétiques [11].
 |
Figure 3. Représentation schématique de 41 mutations graves du gène MTHFR et de 2 polymorphismes particulièrement étudiés. Les 41 mutations spécifiées n’ont été identifiées que dans des familles de patients présentant un déficit important de MTHFR. La protéine MTHFR est représentée par un rectangle et les acides aminés mutés sont indiqués au-dessus, ainsi que les mutations affectant l’épissage du gène. Les nombres sous le rectangle désignent la position des mutations dans la séquence de l’ADNc (GenBank GI:6174884); 2 mutations différentes ont été identifiées à la position 1274. Les nombres entre parenthèses désignent la présence d’une mutation intronique à proximité du résidu mentionné. Les 2 polymorphismes de MTHFR les plus étudiés sont aussi indiqués, en vert. Les mutations 1755G → A (M581I) et 1793G → A (R594Q) ne sont pas représentées ici car leur effet reste ambigu. Elles ont initialement été identifiées chez des patients présentant un défaut prononcé de la MTHFR [8, 9], mais Martin et al. [10] ont suggéré qu’il s’agit de polymorphismes plus ou moins communs selon la population analysée. La mutation 459C → T (indiquée par « * ») est muette et elle est représentée ici à titre indicatif car elle accompagne exclusivement la mutation 458G → T (G149V, surmontée par « * »). |
Études moléculaires et biochimiques du polymorphisme 677C → T
Le polymorphisme C → T à la pb 677 a été identifié de manière paradoxale en examinant un patient souffrant d’une altération sévère de MTHFR [5]. De fait, ce polymorphisme se retrouve autant dans les groupes témoins que chez les patients homocystinuriques, bien que ces derniers soient en plus victimes de mutations graves. La substitution 677 C → T met en jeu une enzyme thermolabile possédant une activité moindre, c’est pourquoi le génotype mutant homozygote (677TT) est associé à une légère hyperhomocystéinémie [5]. Cette association est encore plus évidente lorsque l’on considère des individus dont le sang présente un taux de folates réduit [12]. Cela suggère qu’un simple supplément en acide folique est efficace pour traiter l’hyperhomocystéinémie chez les individus affectés par ce polymorphisme.
Les études biochimiques des versions 677C et 677T de l’enzyme ont fourni une explication à l’effet protecteur de l’acide folique sur l’hyperhomocystéinémie des individus mutants. Des extraits de lymphocytes conservent une activité résiduelle plus élevée après avoir été chauffés si l’on augmente la quantité de folate ; l’effet protecteur est meilleur pour l’enzyme mutante que pour l’enzyme normale [13]. Ce phénomène a été reproduit en chauffant des extraits de bactéries qui expriment l’enzyme recombinante normale ou mutante, ainsi qu’en utilisant l’enzyme humaine purifiée (chapitre 3 de [1]). Le FAD (flavin adenine dinucleotide), cofacteur de la MTHFR, protège l’enzyme mutante de la déstabilisation ; par conséquent, la riboflavine (vitamine B2, précurseur du FAD) peut être considérée comme un déterminant de son activité enzymatique et de l’hyperhomocystéinémie (chapitre 6 de [1]). Le variant 677C→T accroît la tendance de l’enzyme à perdre le FAD, et les folates protègent l’enzyme en atténuant cette perte. Par conséquent, bien que l’identité des mutations sévères soit un bon indicateur de l’activité enzymatique résiduelle des patients homocystinuriques, le polymorphisme (et des facteurs externes comme l’acide folique ou la riboflavine) contribue à la complexité de la relation génotype-phénotype (chapitre 1 de [1]).
La multitude de répercussions cliniques du polymorphisme 677C → T
Le produit de la réaction catalysée par la MTHFR est le 5-méthyltétrahydrofolate (Figure 1). Son seul rôle connu se manifeste par le transfert de son groupe méthyle lors de la reméthylation de l’homocystéine par la méthionine synthase. La MTHFR peut ainsi moduler les taux d’homocystéine, de méthionine et de S-adénosylméthionine. La MTHFR peut aussi indirectement influencer les taux de nucléotides, puisque son substrat, le 5,10-méthylène tétrahydrofolate, est utilisé pour la synthèse de la thymidine et qu’il peut aussi être converti en d’autres dérivés foliques qui participent à la synthèse des purines. La Figure 4 montre la prévalence de l’homozygotie de l’allèle 677T par régions. Heureusement, l’interaction entre les prédispositions génétiques (677TT) et des paramètres non-génétiques (acide folique alimentaire, suppléments vitaminés) est susceptible de prévenir certaines manifestations cliniques associées à une légère déficience en MTHFR.
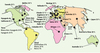 |
Figure 4. Fréquence des homozygotes 677TT pour différentes régions géographiques. L’origine ethnique est aussi indiquée pour certaines populations. L’essentiel de ces données a préalablement été discuté (chapitre 1 de [1]). Les données pour l’Inde et la Grèce sont dérivées de Herrmann et al. [14] et Zalavras et al. [15], respectivement. |
Bien que les mécanismes par lesquels ce polymorphisme influence la progression de différentes maladies peuvent être uniques à chacune de celles-ci, les différentes possibilités incluent (chapitre 1 de [1]) :
Une augmentation des taux d’homocystéine, ce qui peut avoir des effets sur la vascularisation ou le développement des embryons [16].
La perturbation de la synthèse de la méthionine et de la S-adénosylméthionine, avec des répercussions sur la synthèse des protéines ou les réactions de méthylation. De plus, l’homocystéine peut être convertie en S-adénosylhomocystéine, un inhibiteur de plusieurs méthyltransférases. Puisque des changements de méthylation de l’ADN affectent l’expression des gènes, une déficience de MTHFR peut influencer l’amorce puis le développement de processus oncogéniques [17].
Une redistribution des métabolites du folate. Cela peut affecter la synthèse des purines et des pyrimidines (Figure 1), avec des conséquences sur la synthèse ou la réparation de l’ADN.
Toute maladie multifactorielle associée à l’hyperhomocystéinémie peut être influencée par le variant 677T. Malgré la multiplicité des activités de recherche consacrée à ce polymorphisme (Figure 2), un nombre limité de travaux sont cités ici. C’est pourquoi nous invitons le lecteur à consulter des revues récentes [1, 18].
-
Problèmes vasculaires
Puisque l’hyperhomocystéinémie constitue un facteur qui se conjugue aux autres facteurs en cause dans les maladies cardiovasculaires, le variant 677C→T peut modifier la susceptibilité des sujets à l’égard de ces maladies qui ont pour caractéristique d’être multifactorielles. Des méta-analyses suggèrent que le génotype 677TT peut être un facteur de risque modeste en ce qui concerne les accidents vasculaires cérébraux, la thrombose veineuse et les maladies coronariennes, spécialement s’il y a une carence en acide folique (chapitres 8 et 9 de [1]).
-
Défauts du tube neural et autres anomalies congénitales
Quelques rapports [19, 20] ont fait état de taux élevés d’homocystéine plasmatique totale dans des familles présentant des affections du tube neural. Un supplément en folates réduit l’incidence des défauts du tube neural. Le variant 677T est le premier facteur de risque génétique qui a été proposé comme source de cette anomalie [21]. Le risque est environ 2 fois plus grand si l’enfant ou la mère possède le génotype 677TT (chapitre 10 de [1]). Des analyses préliminaires suggèrent qu’il existe une association entre le génotype 677T et l’occurrence des fentes palatines ou d’anomalies congénitales cardiaques, mais un plus grand nombre d’études serait nécessaire afin de confirmer ces observations.
-
Complications de la grossesse
Une méta-analyse a déterminé que le génotype 677TT augmente le taux d’avortement spontané de 30 %-40 %, l’effet étant plus prononcé en début de grossesse. Le génotype 677TT augmente de 40 % le risque de problèmes vasculaires placentaires et de 20 % la probabilité de pré-éclampsie (chapitre 11 de [1]).
-
Troubles neuropsychiatriques
Une association a été notée entre la schizophrénie et l’allèle 677T. D’autres études ont produit des résultats mitigés, probablement à cause de l’hétérogénéité des cas de schizophrénie (chapitre 12 de [1]). Une analyse, qui considérait 6 000 individus, a indiqué que le génotype 677TT cause une augmentation de 70 % du risque de dépression [18]. Des travaux sur la démence et le déclin cognitif léger ont permis de constater qu’il existe une augmentation des taux d’homocystéine plasmatique chez un nombre significatif de patients [18]. Puisque la démence est caractérisée, entre autres, par un affaiblissement des vaisseaux sanguins qui irriguent le cerveau, le variant 677T représente donc un facteur de risque potentiel. Le lien entre le polymorphisme et la démence d’origine vasculaire ou encore la maladie d’Alzheimer n’a toutefois pas été clairement établi (chapitre 12 de [1]).
-
Cancer
Un apport insuffisant en folates prédispose au cancer colorectal. Cependant, si l’apport en acide folique est suffisant, le génotype 677TT est paradoxalement associé à une diminution du risque de cancer colorectal. Il exerce aussi un effet protecteur dans le cas des leucémies lymphocytaires aiguës [18]. Le mécanisme proposé pour expliquer ce phénomène tient à une redistribution des folates, facilitant ainsi la synthèse de la thymidine et réduisant les risques de dommage à l’ADN. Lorsque l’apport en folates est insuffisant, l’augmentation du risque de contracter certains cancers est associée au génotype TT [22].
Autres polymorphismes
Une soixantaine de polymorphismes de MTHFR ont été identifiés [10]. Les substitutions aux bases 677 et 1298 (Figure 3) ont un effet sur l’activité de l’enzyme (chapitre 1 de [1]), mais le mutant 1298A → C n’a fait l’objet que d’un nombre restreint d’études. L’enzyme mutée à la base 1298 n’est pas thermolabile. En présence de 1298A → C (E429A), l’activité de l’enzyme est diminuée de 30 % ; en comparaison, l’allèle 677T en décroît l’activité d’environ 55 %. Les allèles 677T et 1298C sont rarement en configuration cis (chapitre 1 de [1]). Les répercussions fonctionnelles des autres polymorphismes ont été peu caractérisées ou demeurent inconnues.
Modèle animal
Nous avons produit des souris dont le gène Mthfr est inactivé [23]. Les taux d’homocystéine plasmatique totale de ces souris hétérozygotes et homozygotes sont respectivement 1, 6 et 10 fois supérieurs à ceux des témoins sains testés. Les hétérozygotes Mthfr+/- comportent un phénotype normal. De plus petite taille, les homozygotes présentent des retards de développement, ainsi que des pathologies cérébelleuses et hépatiques. On a observé des dépôts lipidiques anormaux dans l’aorte proximale des hétérozygotes plus âgés et des homozygotes. La souris Mthfr−/− constitue un bon modèle pour étudier l’homocystinurie causée par un déficit prononcé en MTHFR. Elle présente, en effet, l’intérêt de comporter des caractéristiques similaires à celles des humains qui sont homozygotes pour 677T.
L’expression de plusieurs gènes du cerveau des souris Mthfr−/− est altérée [24] et une augmentation de l’apoptose a été observée dans le cervelet [25]. Ces souris présentent une stéatose hépatique qui pourrait être reliée à la diminution des taux de métabolites de la choline dans le foie [26].
Ces souris ont servi à tester l’effet de la bétaïne dans les cas de déficience en MTHFR [26]. On a noté que des suppléments de bétaïne réduisent leur homocystéinémie, améliorent leur structure cérébelleuse et limitent leur stéatose hépatique. Ces résultats valorisent le choix de la bétaïne à titre de donneur alternatif de groupe méthyle. D’ailleurs, on a observé une corrélation négative entre les taux plasmatiques d’homocystéine et les taux de bétaïne des patients victimes d’une maladie cardiovasculaire, indiquant que la bétaïne est métabolisée afin de réduire les taux d’homocystéine.
Considéré sous un autre angle, le modèle murin a permis d’observer qu’une insuffisance de Mthfr diminue l’expression de apoA-1, la principale protéine des HDL, composé du cholestérol ; cette observation a été étendue aux humains hyperhomocysteinémiques, décelant ainsi un nouveau mécanisme par lequel un déficit en MTHFR peut accroître le risque d’apparition d’affections vasculaires [27]. Par ailleurs, les souris porteuses d’une légère déficience en MTHFR et/ou ayant subi un régime pauvre en folates connaissent une augmentation de l’incidence des avortements, des retards de croissance intra-utérine et des défauts cardiaques [28]. Ces faits concordent avec les résultats d’études suggérant que le génotype 677T et/ou un apport insuffisant en folates favorisent des complications de la grossesse ainsi que des anomalies congénitales. Finalement, les souris développent des tumeurs intestinales quand on les soumet à un régime alimentaire pauvre en folates [29]. Le nombre de tumeurs est plus élevé si les souris montrent de manière concomitante une déficience légère en MTHFR : ce phénomène suggère que les deux facteurs concourent à accroître le risque d’apparition de tumeurs intestinales.
Conclusions
Depuis l’identification de l’ADNc de MTHFR, les travaux qui se sont déroulés au cours des dix récentes années ont remarquablement développé les connaissances au sujet du gène MTHFR, ses variations de séquence, ainsi que son rôle dans diverses maladies. Cependant, plusieurs aspects importants demeurent peu explorés. Les travaux doivent se poursuivre afin de mieux identifier les régions régulatrices du gène et de comprendre leur modulation [7], ainsi que les facteurs qui influencent l’épissage alternatif de ses ARNm et la synthèse de ses deux isoformes protéiques [6]. Enfin, la régulation de l’enzyme par phosphorylation constitue un nouveau domaine à éclaircir [30].
La prochaine décennie permettra probablement d’évaluer les répercussions cliniques du polymorphisme sur d’autres maladies. Il est certes possible que 677C → T ne contribue que modestement à l’émergence de certaines pathologies, mais il est également possible qu’en étudiant l’interaction du polymorphisme avec d’autres facteurs génétiques, ainsi qu’avec des facteurs non-génétiques, on puisse élucider la cause de pathologies complexes [31]. Enfin, l’aspect pharmacogénétique de 677C → T n’a été qu’effleuré jusqu’à maintenant [18, 32, 33]. Tous ces sujets promettent de voir surgir de fascinants projets de recherche dans la mesure où ils prendront en considération la complexité du gène et les activités métaboliques de MTHFR.
Article reçu le 19 octobre 2006, accepté le 24 janvier 2007.
Remerciements
Les auteurs remercient les IRSC (Instituts de recherche en santé du Canada) pour leur soutien financier. Les auteurs tiennent également à remercier Nada Jabado, Hélène Ammann et Gaston Lalumière pour leur relecture attentive du manuscrit et leurs judicieux conseils.
Références
- Ueland PM, Rozen R (eds). MTHFR polymorphisms and disease. Georgetown: Landes Bioscience/Eurekah.com, 2005 : 210 p. [Google Scholar]
- Mudd SH, Uhlendorf BW, Freeman JM, et al. Homocystinuria associated with decreased methylenetetrahydrofolate reductase activity. Biochem Biophys Res Commun 1972; 46 : 905–12. [Google Scholar]
- Kang SS, Zhou J, Wong PW, et al. Intermediate homocysteinemia: a thermolabile variant of methylenetetrahydrofolate reductase. Am J Hum Genet 1988; 43 : 414–21. [Google Scholar]
- Goyette P, Sumner JS, Milos R, et al. Human methylenetetrahydrofolate reductase: isolation of cDNA, mapping and mutation identification. Nat Genet 1994; 7 : 195–200. [Google Scholar]
- Frosst P, Blom HJ, Milos R, et al. A candidate genetic risk factor for vascular disease: a common mutation in methylenetetrahydrofolate reductase. Nat Genet 1995; 10 : 111–3. [Google Scholar]
- Tran P, Leclerc D, Chan M, et al. Multiple transcription start sites and alternative splicing in the methylenetetrahydrofolate reductase gene result in two enzyme isoforms. Mamm Genome 2002; 13 : 483–92. [Google Scholar]
- Pickell L, Tran P, Leclerc D, et al. Regulatory studies of murine methylenetetrahydrofolate reductase reveal two major promoters and NF-kappaB sensitivity. Biochim Biophys Acta 2005; 1731 : 104–14. [Google Scholar]
- Selzer RR, Rosenblatt DS, Laxova R, Hogan K. Adverse effect of nitrous oxide in a child with 5,10-methylenetetrahydrofolate reductase deficiency. N Engl J Med 2003; 349 : 45–50. [Google Scholar]
- Tonetti C, Saudubray JM, Echenne B, et al. Relations between molecular and biological abnormalities in 11 families from siblings affected with methylenetetrahydrofolate reductase deficiency. Eur J Pediatr 2003; 162 : 466–75. [Google Scholar]
- Martin YN, Salavaggione OE, Eckloff BW, et al. Human methylenetetrahydrofolate reductase pharmacogenomics: gene resequencing and functional genomics. Pharmacogenet Genomics 2006; 16 : 265–77. [Google Scholar]
- Morel CF, Scott P, Christensen E, et al. Prenatal diagnosis for severe methylenetetrahydrofolate reductase deficiency by linkage analysis and enzymatic assay. Mol Genet Metab 2005; 85 : 115–20. [Google Scholar]
- Jacques PF, Bostom AG, Williams RR, et al. Relation between folate status, a common mutation in methylenetetrahydrofolate reductase, and plasma homocysteine concentrations. Circulation 1996; 93 : 7–9. [Google Scholar]
- Guenther BD, Sheppard CA, Tran P, et al. The structure and properties of methylenetetrahydrofolate reductase from Escherichia coli suggest how folate ameliorates human hyperhomocysteinemia. Nat Struct Biol 1999; 6 : 359–65. [Google Scholar]
- Herrmann FH, Salazar-Sanchez L, Schröder W, et al. Prevalence of molecular risk factors FV Leiden, FV HR2, FII 20210G>A and MTHFR 677C > T in different populations and ethnic groups of Germany, Costa Rica and India. Indian J Hum Genet 2001; 1 : 33–9. [Google Scholar]
- Zalavras CG, Giotopoulou S, Dokou E, et al. Lack of association between the C677T mutation in the 5,10-methylenetetrahydrofolate reductase gene and venous thromboembolism in Northwestern Greece. Int Angiol 2002; 21 : 268–71. [Google Scholar]
- Junien C, Gallou-Kabani C, Vigé A, Gross MS. Épigénomique nutritionelle du syndrome métabolique. Med Sci (Paris) 2005; 21 : 396–404. [Google Scholar]
- Deltour S, Chopin V, Leprince D. Modifications épigénétiques et cancer. Med Sci (Paris) 2005; 21 : 405–11. [Google Scholar]
- Rozen R. Methylenetetrahydrofolate reductase gene polymorphism - clinical implications. In : Fuchs J, Podda M, eds. Encyclopedia of medical genomics and proteomics. New York: Taylor and Francis Group, 2005. DOI: 10.1081/E-EDGP-120030861. [Google Scholar]
- Mills JL, McPartlin JM, Kirke PN, et al. Homocysteine metabolism in pregnancies complicated by neural-tube defects. Lancet 1995; 345 : 149–51. [Google Scholar]
- Steegers-Theunissen RP, Boers GH, Blom HJ, et al. Neural tube defects and elevated homocysteine levels in amniotic fluid. Am J Obstet Gynecol 1995; 172 : 1436–41. [Google Scholar]
- Van der Put NMJ, Steegers-Theunissen RPM, Frosst P, et al. Mutated methylenetetrahydrofolate reductase as a risk factor for spina bifida. Lancet 1995; 346 : 1070–1. [Google Scholar]
- Heijmans BT, Boer JM, Suchiman HE, et al. A common variant of the methylenetetrahydrofolate reductase gene (1p36) is associated with an increased risk of cancer. Cancer Res 2003; 63 : 1249–53. [Google Scholar]
- Chen Z, Karaplis AC, Ackerman SL, et al. Mice deficient in methylenetetrahydrofolate reductase exhibit hyperhomocysteinemia and decreased methylation capacity, with neuropathology and aortic lipid deposition. Hum Mol Genet 2001; 10 : 433–43. [Google Scholar]
- Chen Z, Ge B, Hudson TJ, Rozen R. Microarray analysis of brain RNA in mice with methylenetetrahydrofolate reductase deficiency and hyperhomocysteinemia. Brain Res Gene Expr Patterns 2002; 1 : 89–93. [Google Scholar]
- Chen Z, Schwahn BC, Wu Q, et al. Postnatal cerebellar defects in mice deficient in methylenetetrahydrofolate reductase. Int J Dev Neurosci 2005; 23 : 465–74. [Google Scholar]
- Schwahn BC, Chen Z, Laryea MD, et al. Homocysteine-betaine interactions in a murine model of 5,10-methylenetetrahydrofolate reductase deficiency. FASEB J 2003; 17 : 512–4. [Google Scholar]
- Mikael LG, Genest J Jr, Rozen R. Elevated homocysteine reduces apolipoprotein A-I expression in hyperhomocysteinemic mice and in males with coronary artery disease. Circ Res 2006; 98 : 564–71. [Google Scholar]
- Li D, Pickell L, Liu Y, et al. Maternal methylenetetrahydrofolate reductase deficiency and low dietary folate lead to adverse reproductive outcomes and congenital heart defects in mice. Am J Clin Nutr 2005; 82 : 188–95. [Google Scholar]
- Knock E, Deng, L, Wu Q, et al. Low dietary folate initiates intestinal tumours in mice, with altered expression of G2M checkpoint regulators Plk1 and Cdc25c. Cancer Res 2006; 66 : 10341–56. [Google Scholar]
- Yamada K, Strahler JR, Andrews PC, Matthews RG. Regulation of human methylenetetrahydrofolate reductase by phosphorylation. Proc Natl Acad Sci USA. 2005; 102 : 10454–9. [Google Scholar]
- Turleau C, Vekemans M. Nouvelles données en génétique chromosomique. Med Sci (Paris) 2005; 21 : 940–6. [Google Scholar]
- Cohen V, Panet-Raymond V, Sabbaghian N, et al. Methylenetetrahydrofolate reductase polymorphism in advanced colorectal cancer: a novel genomic predictor of clinical response to fluoropyrimidine-based chemotherapy. Clin Cancer Res 2003; 9 : 1611–5. [Google Scholar]
- Kim YI. 5,10-Methylenetetrahydrofolate reductase polymorphisms and pharmacogenetics: a new role of single nucleotide polymorphisms in the folate metabolic pathway in human health and disease. Nutr Rev 2005; 63 : 398–407. [Google Scholar]
Liste des figures
|
Figure 1. Répercussions métaboliques de l’activité de la MTHFR. La 5,10-méthylène tétrahydrofolate réductase (MTHFR) catalyse la réduction irréversible du 5,10-methylène tétrahydrofolate (CH2THF) en 5-méthyltétrahydrofolate (CH3THF). L’activité de la MTHFR affecte ainsi la disponibilité du CH2THF, ce qui influence la synthèse de l’ARN et de l’ADN. Le CH3THF est requis pour la reméthylation de l’homocystéine (Hcy) en méthionine (MET), qui intervient elle-même dans la synthèse protéique et la méthylation de l’ADN et d’autres composés (CH3-X). Le FAD est le cofacteur de la MTHFR. MS, méthionine synthase ; TS, thymidylate synthase ; THF, tétrahydrofolate ; DHF, dihydrofolate ; CHOTHF, 10-formyltétrahydrofolate ; SAM, S-adénosyl méthionine. |
| Dans le texte | |
|
Figure 2. Fréquence annuelle des publications traitant de la MTHFR entre 1988 et 2005. Le nombre d’articles scientifiques discutant de la MTHFR (cercles bleus) a été estimé en interrogeant la plate-forme Ovid MEDLINE à l’aide des termes « (MTHFR OR methylenetetrahydrofolate reductase) ». Les articles énonçant plus spécifiquement des mutations de MTHFR ont été identifiés en utilisant les mots-clés « (MTHFR OR methylenetetrahydrofolate reductase) AND (polymorphism OR 677 OR C677T OR variant OR mutation OR genotype OR risk factor OR thermolabile) » (carrés rouges). L’examen sommaire d’un échantillon représentatif de ces publications montre que la quasi-totalité d’entre elles mentionnent le polymorphisme 677C → T. |
| Dans le texte | |
|
Figure 3. Représentation schématique de 41 mutations graves du gène MTHFR et de 2 polymorphismes particulièrement étudiés. Les 41 mutations spécifiées n’ont été identifiées que dans des familles de patients présentant un déficit important de MTHFR. La protéine MTHFR est représentée par un rectangle et les acides aminés mutés sont indiqués au-dessus, ainsi que les mutations affectant l’épissage du gène. Les nombres sous le rectangle désignent la position des mutations dans la séquence de l’ADNc (GenBank GI:6174884); 2 mutations différentes ont été identifiées à la position 1274. Les nombres entre parenthèses désignent la présence d’une mutation intronique à proximité du résidu mentionné. Les 2 polymorphismes de MTHFR les plus étudiés sont aussi indiqués, en vert. Les mutations 1755G → A (M581I) et 1793G → A (R594Q) ne sont pas représentées ici car leur effet reste ambigu. Elles ont initialement été identifiées chez des patients présentant un défaut prononcé de la MTHFR [8, 9], mais Martin et al. [10] ont suggéré qu’il s’agit de polymorphismes plus ou moins communs selon la population analysée. La mutation 459C → T (indiquée par « * ») est muette et elle est représentée ici à titre indicatif car elle accompagne exclusivement la mutation 458G → T (G149V, surmontée par « * »). |
| Dans le texte | |
|
Figure 4. Fréquence des homozygotes 677TT pour différentes régions géographiques. L’origine ethnique est aussi indiquée pour certaines populations. L’essentiel de ces données a préalablement été discuté (chapitre 1 de [1]). Les données pour l’Inde et la Grèce sont dérivées de Herrmann et al. [14] et Zalavras et al. [15], respectivement. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.