")
")
| Issue |
Med Sci (Paris)
Volume 23, Mars 2007
Protéomique clinique en oncologie
|
|
|---|---|---|
| Page(s) | 19 - 22 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/2007231s19 | |
| Published online | 15 mars 2007 | |
Vers une standardisation des outils pour les études de protéomique clinique
Towards a standardization of the tools for the studies of clinical proteomics
Plate-forme Protéomique IFR-Santé-STIC, 8, boulevard de Lattre de Tassigny, 21000 Dijon, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
L’identification de marqueurs protéiques d’intérêt diagnostique ou pronostique a suscité de grands espoirs en oncologie. À l’heure actuelle, cependant, la protéomique clinique fondée sur l’analyse de profils protéiques dans les liquides biologiques se heurte à un défaut de sensibilité et de reproductibilité. Cela est dû au manque de standardisation des étapes pré-analytiques et analytiques. La taille de la cohorte de patients analysés est un autre paramètre essentiel pour donner de la puissance à l’analyse envisagée. Le recrutement d’une cohorte suffisante justifie les études multicentriques qui imposent l’analyse de la reproductibilité inter-plates-formes et la standardisation des étapes pré-analytiques et analytiques. Cette standardisation passe par la mise en place d’échantillons de référence.
Abstract
Proteins identified in biological fluids of cancer patients could be helpful for both diagnosis and prognosis. However, clinical proteomics based on analysis of protein profiles in biological fluids has demonstrated various flaws, most of them related to the difficulties met in reproducibility. These difficulties could be partly overcome by accurate standardisation of pre-analytical and analytical steps of these studies. The size of the patient cohort is one of the parameters that determine the powerfulness of the study. Recruitment of a cohort with a sufficient size often implies multicentric studies in which analysis of the reproducibility between centres and standardisation of pre-analytical and analytical steps are essential. Such a standardisation requires the use of calibrated samples as common references.
© 2007 médecine/sciences - Inserm / SRMS
Dans les laboratoires d’analyse médicale de routine, l’identification et le dosage de protéines connues se font selon des modes opératoires bien définis permettant de comparer les valeurs obtenues à des valeurs de référence considérées comme normales. Ces modes opératoires répondent à une exigence : l’assurance qualité. Les protocoles et modes opératoires qui régissent la recherche de nouveaux biomarqueurs par une analyse protéomique du sérum ou d’autres liquides biologiques ne sont pas encore bien définis. Cette recherche, fondée sur l’analyse de profils protéiques engendrés par spectrométrie de masse, comporte de nombreuses étapes pré-analytiques (prélèvement, conservation, traitement des échantillons), analytiques (spectrométrie de masse) et post-analytiques (stockage, traitement et analyse des données engendrées). Ces étapes doivent s’appliquer à un nombre suffisant d’échantillons pour que les informations fournies soient suffisamment robustes. Après une période de mise au point et d’évolution technologique, il est maintenant indispensable de standardiser les protocoles et les modes opératoires afin de s’assurer de la qualité des données produites [1, 2]. La définition de la taille de l’échantillon étudié participe à la qualité de cette approche.
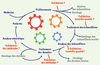 |
Figure 1. Enchaînement des étapes préanalytiques et analytiques nécessaires dans le cadre de la recherche de biomarqueurs en protéomique clinique. Points de contrôle qualité nécessaires à mettre en place. |
 |
Figure 2. Schéma des solutions de l’analyse multicentrique des sérums et/ou plasmas à partir d’un recrutement multicentrique. |
L’importance de la taille de l’échantillon
L’une des clés du succès de ces nouvelles approches est l’identification et l’optimisation de modèles statistiques adaptés à la comparaison de profils protéiques complexes. On n’est pas ici dans une situation où chaque protéine étudiée peut être considérée comme une variable indépendante. C’est l’analyse du profil d’expression d’une ou plusieurs dizaines de peptides qui permettra de discriminer plusieurs populations de patients (sains/malades ; sensibles/résistants à un traitement donné ; de bon pronostic/de mauvais pronostic ; forme clinique A/forme clinique B). Cette analyse s’apparente donc à celles utilisées pour les études du transcriptome. L’analyse statistique des données issues de l’étude des puces à ADN a démontré la fragilité des signatures géniques prédictives. Il suffit de faire varier la taille de l’échantillon pour modifier cette signature. Il est classique d’identifier une signature sur une première série d’échantillons (training) et de la valider sur une seconde série en utilisant la même technique. La variation de la taille de ces deux sous-groupes peut suffire à faire varier la signature génique prédictive comme cela a été montré en reprenant les données de 7 études publiées dans les lymphomes malins non hodgkiniens, les carcinomes hépatocellulaires, les cancers du sein, les médulloblastomes ou les adénocarcinomes pulmonaires [3–5]. Un des paramètres permettant de stabiliser la signature génique prédictive est l’augmentation de la taille de l’étude [5].
La multiplication d’études de taille insuffisante ou mal calibrées, en produisant des données peu reproductibles, participe à la déception engendrée, après une phase d’engouement excessif, par ces approches lourdes et délicates. Des contrôles internes et externes doivent permettre de vérifier que l’interprétation des résultats ne pêche pas par excès d’optimisme.
La dimension multicentrique des études
Les études de petites tailles ont l’avantage de permettre d’effectuer les étapes pré-analytiques et analytiques au sein d’un même laboratoire. L’augmentation de la taille des études, nécessaire à l’identification de profils protéiques d’intérêt diagnostique ou pronostique, nécessite de mettre en place des études multicentriques, seules capables de recruter un nombre suffisant d’échantillons biologiques dans un délai raisonnable [5].
Cette dimension multicentrique impose une standardisation particulièrement soigneuse des étapes pré-analytiques, incluant le transport des échantillons si l’analyse est réalisée dans un centre unique, et des étapes analytiques lorsque l’étude est réalisée en parallèle dans plusieurs centres. Dans cette dernière hypothèse, l’analyse doit être validée par les différents laboratoires sur plusieurs séries d’échantillons successives pendant une période relativement courte. Ceci a le mérite de mettre en place de véritables réseaux qui devraient permettre d’analyser plus rapidement et plus efficacement des séries de patients de taille significative.
Afin de limiter la variabilité inter-plates- formes, les procédures doivent être homogénéisées, ce qui nécessite une concertation des laboratoires impliqués afin de définir des procédures communes, de standardiser l’utilisation des instruments et d’identifier les outils de référence à chaque étape du traitement et de l’analyse de l’échantillon.
Pour chaque étude, l’analyse statistique des données peut être centralisée et traitée par une équipe dédiée. Il est essentiel que l’équipe qui fera cette analyse statistique soit consultée dès la mise en place de l’étude. Elle doit contribuer à la formulation de l’hypothèse de travail, valider le protocole expérimental et la taille de la cohorte étudiée et indiquer toutes les informations qu’il est nécessaire de recueillir pour l’analyse ultérieure.
Trois étapes sont nécessaires pour standardiser une analyse multicentrique : (1) la standardisation instrumentale, validée par la reproductibilité de la mesure réalisée avec divers instruments sur un échantillon simple ; (2) la définition concertée de modes et/ou procédures opératoires communes ; (3) le choix d’un échantillon de référence (ou standard) permettant de calibrer régulièrement ces analyses et de valider chaque expérience.
La standardisation instrumentale
La standardisation instrumentale doit être effectuée à partir d’un échantillon simple qui sert de contrôle qualité de l’instrument de mesure et valide l’étape analytique. Cette standardisation a été réalisée par la société Ciphergen dans une série d’études réalisées aux États- Unis puis en Europe [7, 8]. La société Bruker Daltonique mesure actuellement la variabilité instrumentale d’un site à l’autre, en France (Lyon, Dijon, Marseille, Wissembourg) et en Allemagne (Leipzig, Bremen). Cette standardisation permet d’adapter un certain nombre de paramètres techniques [9], aussi bien au niveau hardware que des méthodes d’acquisition automatique, afin d’assurer la reproductibilité inter-centres des mesures tout en conservant la meilleure sensibilité possible. Les paramètres pris en compte sont la résolution, le rapport signal sur bruit et l’intensité du signal.
Cette procédure de standardisation instrumentale concerne actuellement 7 spectromètres de masse de type MALDI-TOF (3 autoflex et 4 ultraflex, Bruker Daltonique). L’échantillon de référence utilisé pour cette étude est un mélange de deux calibrants (protéine I et peptide calibrant standard). Chaque laboratoire utilise en effet cet échantillon de référence en routine pour la calibration de la mesure de masse. Dans un premier temps, le même échantillon a été analysé par différentes plates-formes de protéomique. Puis les paramètres d’acquisition ont été revus afin d’homogénéiser les résultats. Chaque laboratoire analyse maintenant de manière régulière cet échantillon de référence afin de vérifier la stabilité des données engendrées par les instruments.
En cours d’étude, l’échantillon témoin permet de détecter une éventuelle dérive instrumentale et de corriger si nécessaire les paramètres d’acquisition du spectromètre de masse. Les résultats de ce contrôle, appliqué avant toute analyse d’échantillon biologique, doivent être conservés et tracés.
Les procédures opératoires communes et la calibration régulière
Le traitement des échantillons (purification, déplétion des protéines abondantes, fractionnement) doit se faire selon des procédures opératoires pré-définies. Ces procédures opératoires n’empêchent pas la nécessité d’une validation. Cette validation est un contrôle de qualité. Elle est fondée sur l’utilisation d’un échantillon de référence qui permet d’abord de mesurer les variations inter-laboratoires et de mettre en place des actions correctives. Si les variations inter-laboratoires sont faibles, l’échantillon de référence permet de neutraliser les variations intra-laboratoires (par exemple, une dérive expérimentale provenant de changements de lot de consommables) par une normalisation des résultats par rapport à cet échantillon.
Nous avons développé, en collaboration avec l’Établissement Français du Sang Bourgogne-Franche Comté, un échantillon de référence (CQI) qui permet de valider les modes opératoires au sein de la plate-forme protéomique de Dijon. Ce CQI correspond à un pool de plasma inactivé, conditionné en 2 000 paillettes, dans des conditions contrôlées. Ces paillettes contiennent 450 μl d’échantillon, sont scellées à leurs deux extrémités et sont conservées à − 196°C. Afin de vérifier leur homogénéité, trois lots des paillettes prélevées en début, milieu et fin de processus ont été analysés. Après purification de sous-protéomes à l’aide de billes magnétiques (MB-HIC 18 et MB-WCX, Bruker Daltonique), des profils protéiques sur la gamme de masse 1 000- 10 000 ont été engendrés. La Figure 3 montre que les profils protéiques obtenus à partir des différents lots de paillettes sont sensiblement identiques.
 |
Figure 3. Profil protéomique moyen obtenu après traitement (purification par billes HIC-C18) et analyse de trois lots de paillettes servant d’échantillons de références et confectionnées en début, milieu et fin de processus. arb. u. : unités arbitraires. |
Cet échantillon de référence CQI est aujourd’hui utilisé par plusieurs laboratoires dans le cadre de la standardisation. Pour cela, chaque laboratoire suit le même protocole de traitement (purification par billes magnétiques de types MB-HIC 18 et MB-WCX) et d’analyse des échantillons. Les échantillons analysés sont l’échantillon de référence CQI et le mélange peptide-protéine utilisé pour la calibration instrumentale.
Cette étape est essentielle au moment où l’on cherche à accroître la sensibilité des méthodes de détection de nouveaux bio-marqueurs en utilisant le fractionnement de l’échantillon, la déplétion des protéines abondantes, la purification d’un sous-protéome spécifique (phosphorylation, glycosilation, etc.) [10–12]. La reproductibilité de ces méthodes, lesquelles comportent parfois plus de 20 étapes différentes, reste mal connue. Le CQI est utilisé pour : (1) analyser le niveau de reproductibilité de ces méthodes ; (2) identifier les étapes critiques, celles susceptibles d’engendrer une forte variabilité ; (3) définir si ces méthodes sont applicables à des analyses multicentriques à grande échelle.
Dans le cadre d’un groupe de travail constitué par l’Inca, un début de réflexion a été initiée en France sur l’importance de standardiser la nomenclature à appliquer sur les données produites dans le domaine de la protéomique clinique. Ceci est indispensable si l’on souhaite une bonne communication entre les laboratoires d’analyses et les équipes de statisticiens qui devront traiter et analyser les données. Cependant, la diversité des procédures de purification et d’analyse des données rend la tâche difficile.
Conclusions
L’utilisation des nouvelles technologies de protéomique clinique impose la mise en place des outils de contrôle nécessaires à la réalisation d’études multicentriques de grande envergure. La standardisation des instruments et des procédures opératoires est réalisable et peut être validée par l’analyse d’un échantillon commun permettant de mesurer la variabilité intra et interlaboratoires. Si celle-ci est trop importante (bruit de fond supérieur à la sensibilité des techniques et des biomarqueurs), il faudra imaginer des approches différentes. Si la variabilité intra et interlaboratoires est correcte, il sera possible d’envisager la constitution de banques de profils protéiques, chaque profil étant accompagné des différents éléments de contrôle qualité (pré-analytique et analytique) ainsi que de l’ensemble des informations concernant l’échantillon (lieu de stockage, mode de conservation, date de prélèvement, lien avec le centre de ressource biologique qui dispose de l’échantillon, pathologie). Cette banque de données permettra à chaque laboratoire de rapidement comparer de nouveaux profils à ceux déjà engendrés selon la même méthode, comme il est possible de comparer des profils expérimentaux à des profils théoriques. Des études comparatives croisées (en fonction d’un critère donné : pathologie, traitement, survie, etc.) permettront de valider rapidement un biomarqueur identifié dans une première étude. L’ensemble de ces mesures devrait permettre d’accroître à moyen terme la fiabilité des résultats tout en diminuant le temps de recrutement et d’analyse ainsi que le coût des études de protéomique clinique.
Références
- Diamandis EP. Mass spectrometry as a diagnostic and a cancer biomarker discovery tool: opportunities and potential limitations. Mol Cell Proteomics 2004; 3 : 367–78. [Google Scholar]
- Baggerly KA, Morris JS, Coombes KR. Reproductibility of SELDI-TOF protein patterns in serum: comparing datasets from different experiments. Bioinformatics 2004; 20 : 777–85. [Google Scholar]
- Tibshirani RJ, Efron B. Pre-validation and inference in microarrays. Stat Appl Genet Mol Biol 2002; 1 : 1–18. [Google Scholar]
- Michiels S, Koscielny S, Hill C. Prediction of cancer outcome with microarrays: a multiple random validation strategy. Lancet 2005; 365 : 488–92. [Google Scholar]
- Anderson NL. The roles of multiple proteomic platforms in a pipeline for new diagnostics. Mol Cell Proteomics 2005; 4 : 1441–4. [Google Scholar]
- Ein-Dor L, Zuk O, Domany E. Thousands of samples are needed to generate a robust gene list for predicting outcome in cancer. Proc Natl Acad Sci USA 2006; 103 : 5923–8. [Google Scholar]
- Semmes OJ, Feng Z, Adam BL, et al. Evaluation of serum protein profiling by surface-enhanced laser desorption/ionization time-of-flight mass spectrometry for the detection of prostate cancer. I. Assessment of platform reproducibility. Clin Chem 2005; 51 : 102–12. [Google Scholar]
- Bons JA, de Boer D, van Dieijen-Visser MP, Wodzig WK. Standardization of calibration and quality control using surface enhanced laser desorptionionization-time of flight-mass spectrometry. Clin Chim Acta 2006; 366 : 249–56. [Google Scholar]
- Baumann S, Ceglarek U, Fiedler GM, et al. Standardized approach to proteome profiling of human serum based on magnetic bead separation and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Clin Chem 2005; 51 : 973–80. [Google Scholar]
- Tang HY, Ali-Khan N, Echan LA, et al. A novel four-dimensional strategy combining protein and peptide separation methods enables detection of low-abundance proteins in human plasma and serum proteomes. Proteomics 2005; 5 : 3329–42. [Google Scholar]
- Echan LA, Tang HY, Ali-Khan N, et al. Depletion of multiple highabundance proteins improves protein profiling capacities of human serum and plasma. Proteomics 2005; 5 : 3292–303. [Google Scholar]
- Yang Z, Harris LE, Palmer-Toy DE, Hancock WS. Multilectin affinity chromatography for characterization of multiple glycoprotein biomarker candidates in serum from breast cancer patients. Clin Chem 2006; 52 :1897–905. [Google Scholar]
Liste des figures
|
Figure 1. Enchaînement des étapes préanalytiques et analytiques nécessaires dans le cadre de la recherche de biomarqueurs en protéomique clinique. Points de contrôle qualité nécessaires à mettre en place. |
| Dans le texte | |
|
Figure 2. Schéma des solutions de l’analyse multicentrique des sérums et/ou plasmas à partir d’un recrutement multicentrique. |
| Dans le texte | |
|
Figure 3. Profil protéomique moyen obtenu après traitement (purification par billes HIC-C18) et analyse de trois lots de paillettes servant d’échantillons de références et confectionnées en début, milieu et fin de processus. arb. u. : unités arbitraires. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.