")
")
| Issue |
Med Sci (Paris)
Volume 22, Number 11, Novembre 2006
|
|
|---|---|---|
| Page(s) | 961 - 968 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/20062211961 | |
| Published online | 15 novembre 2006 | |
Immunité innée antivirale : Rôle des mécanismes Toll-dépendants et Toll-indépendants
Toll-dependent and -independent innate antiviral immunity
1
Laboratoire d’Immunogénétique moléculaire humaine, Centre de recherché en immunologie et hématologie, Faculté de médecine, 4, rue Kirschleger, 67085 Strasbourg Cedex, France
2
Laboratoire central d’immunologie, Hôpitaux universitaires de Strasbourg, Hôpital de Hautepierre, avenue Molière, 67200 Strasbourg, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Jusque récemment, l’immunité adaptative, et plus particulièrement les cellules T cytotoxiques, étaient considérées comme les seuls éléments essentiels dans la défense de l’hôte contre les infections virales. Des données récentes, qui ne remettent toutefois pas en cause le rôle crucial de ces cellules dans ces mécanismes, ont également mis en lumière les fonctions primordiales des cellules de l’immunité innée, macrophages, cellules dendritiques, cellules Tγδ et cellules natural killer (NK), dans la lutte antivirale. En effet, il est maintenant clairement établi que ces cellules sont capables de détecter la présence d’agents infectieux de façon très précoce, par l’intermédiaire de récepteurs spécifiques pour des molécules produites au cours d’infections virales, mais aussi bactériennes, fongiques ou parasitaires. L’engagement de ces récepteurs active alors plusieurs cascades de signalisation qui aboutissent à la mise en place extrêmement rapide de systèmes de défense très efficaces. Par ailleurs, les cytokines synthétisées et libérées au cours de cette première phase de la réponse immunitaire sont essentielles à l’activation des cellules de l’immunité adaptative, qui permettront une lutte spécifique et l’établissement d’une mémoire immunologique contre l’agresseur. Parmi les récepteurs de l’immunité innée antivirale, les récepteurs de la famille Toll (TLR, Toll-like receptors) ont été particulièrement étudiés, et certains d’entre eux sont clairement impliqués dans la défense contre les infections virales. Cependant, de nouvelles molécules, impliquées dans des mécanismes de défense immunitaire antivirale « TLR-indépendants », ont été mises en évidence très récemment et font l’objet d’études approfondies. L’objectif de cet article est de synthétiser les données actuelles concernant la reconnaissance des infections virales par les mécanismes TLR-dépendants et TLR-indépendants, et l’induction de la réponse innée qui en résulte.
Abstract
Until recently, adaptive immunity and cytotoxic T cells were considered as the only essential components of the antiviral defence arsenal. Additional data that do not rule out the crucial role of these cells in the clearance of viral pathogens have, however, recently emerged. They indicate that innate immune cells such as macrophages, dendritic cells, γδ T cells as well as natural killer (NK) cells play a primordial role in this mechanism. It is now well established that innate immune cells can detect various pathogens (bacteria, viruses, fungi or parasites) very rapidly and respond to their presence through the activation of specific receptors. Once activated, these molecules trigger several signalling cascades that culminate in the establishment of very potent defence mechanisms. In addition, cytokines produced during this initial response are essential for the activation of the adaptive immune response which will add specificity and memory to the system. Among the innate immune receptors, attention has focused on the Toll-like receptors (TLR) and many reports indicate that some of the TLRs are clearly involved in defence against viral pathogens. However, new molecules, acting independently from any TLR, have recently been discovered. They define a second antiviral pathway which is presently the subject of intense research. In this article, we will review the role of the different molecules involved in each pathway within the framework of innate antiviral defence.
© 2006 médecine/sciences - Inserm / SRMS
Le système immunitaire des vertébrés est classiquement divisé en deux bras : l’immunité innée et l’immunité adaptative. Cette séparation n’est toutefois pas marquée par une ligne de démarcation nette, car ces deux mécanismes se complémentent et que de nombreux types cellulaires et certaines cytokines interviennent comme médiateurs dans cette interaction [1].
La principale différence entre ces deux bras est que l’activation de l’immunité innée dépend de récepteurs qui ne subissent pas de réarrangements somatiques et qui sont, par conséquent, prédéterminés pour reconnaître des molécules présentes en cas d’infection par différents types de pathogènes (bactéries, virus, parasites ou champignons). Une fois ces récepteurs activés, les cellules qui les expriment (macrophages, cellules dendritiques, cellules NK) agissent directement pour détruire ou limiter la progression de l’agent infectieux. Cette réponse très rapide, débutant dans les minutes qui suivent l’infection, peut être mise en évidence chez tous les représentants du règne animal. En ce sens, il s’agit d’un mécanisme primitif, relativement simple, à reconnaissance directe (pour revue, voir [2]), mais qui ne permet pas l’élaboration d’une mémoire immunologique lorsque l’hôte est à nouveau confronté au même agent infectieux.
À l’inverse, la réponse adaptative, dont l’origine évolutive est plus récente [3], met en œuvre des cellules effectrices (lymphocyte B et T) capables, au terme de collaborations intercellulaires complexes, de produire une réponse spécifiquement dirigée contre l’agresseur. Cette spécificité provient de l’expression de molécules (récepteurs de l’antigène sur les cellules T et production d’anticorps par les cellules B) qui sont le produit de l’expression de gènes ayant subi des réarrangements et de mutations somatiques permettant la production, par l’hôte, d’une très vaste gamme de protéines (le répertoire) potentiellement capable de reconnaître n’importe quel épitope porté par un antigène du non-soi. Une fois activé, ce système complexe conserve la mémoire de l’agresseur, lui permettant ainsi d’y répondre plus rapidement lors d’un contact ultérieur. Cependant, cette réponse requiert plusieurs jours avant d’atteindre son efficacité maximale, ce qui laisse un délai suffisant au pathogène pour se disséminer, surtout si cet agent infectieux a une capacité de multiplication rapide, comme c’est le cas pour certains virus.
Pour limiter la propagation virale dès les premiers signes d’infection, la réponse innée constitue donc une barrière essentielle. Au cours des dernières années, les recherches sur les récepteurs de l’immunité innée se sont focalisées sur les membres de la famille Toll [4, 5] (→)
(→) m/s 2006, n° 2, p. 149.
Dans la plupart des cas, les ligands des différents TLR ont été identifiés [4] et se sont révélés être des molécules d’origine microbienne. Ultérieurement, il a été démontré qu’un certain nombre de TLR sont capables de reconnaître des molécules qui constituent des signatures typiques d’infection virale, comme l’ARN bicaténaire (dans le cas du TLR 3), l’ARN simple brin (pour les TLR 7 et 8 chez l’homme, pour le TLR 7 chez la souris) ou encore d’ADN bicaténaire non méthylé (pour le TLR 9). Avec le TLR 4, qui peut également être activé par des protéines d’origine virale, ces récepteurs (Figure 1A) ont la capacité d’induire la production d’interféron de type I (α/β), ce qui fait d’eux des « détecteurs » d’infection virale de premier choix (pour revue, voir [6]).
 |
Figure 1. Molécules impliquées dans la réponse antivirale TLR-dépendante (A) et TLR-indépendante (B). Différents domaines, identifiés par SMART (simple modular architecture research tool), sont représentés. LRR : régions riches en leucine ; TIR : Toll-interleukin 1 receptor ; TM : domaine transmembranaire ; CARD : domaine de recrutement des caspases ; DEXDc : domaine dead des protéines à hélicase ; DSRM : domaine de liaison à l’ARN double brin ; Lect-C : lectine de type C. La localisation cellulaire de ces protéines est indiquée (RE : réticulum endoplasmique). |
Très récemment, une nouvelle voie de réponse aux infections virale a été mise au jour. Logiquement décrite comme « TLR-indépendante », cette voie de signalisation, qui détecte des molécules d’origine virale comme l’ARN double brin, fait intervenir des récepteurs intracellulaires à domaine hélicase, comme la protéine RIG-I [7, 8], et culmine également avec la production d’interférons de type I. De nombreux travaux sont venus compléter l’étude de cette voie, avec la découverte d’autres composants comme la protéine IPS-1 [9], également appelée MAVS [10], VISA [11] ou Cardif [12]).
Dans cet article, nous nous proposons d’analyser ces données récentes, ainsi que les différents gènes impliqués dans la reconnaissance et la réponse innée antivirale réalisées par des mécanismes TLR-dépendants et TLR-indépendants.
Récepteurs de la famille Toll impliqués dans l’immunité antivirale
TLR 2
Un certain nombre d’études récentes indiquent que TLR 2, classiquement décrit comme un récepteur de lipopeptides di- ou tri-acétylés, répond également à des stimulations d’origine virale (Tableau I) par l’activation de la voie NF-κB. Ces données obtenues in vitro (infections de cellules après transfection transitoire de vecteurs d’expression pour Tlr2) ne permettent pas d’établir le mécanisme par lequel l’activation de TLR2 induit une protection antivirale, mais on peut suggérer que l’induction de l’apoptose des cellules infectées limiterait la dissémination virale. Seule une étude [13] démontre, in vivo, l’importance de TLR 2 dans la résistance des animaux à une infection expérimentale, par voie intrapéritonéale, du virus herpes simplex 1 (HSV-1). De façon surprenante, les auteurs remarquent que l’absence de TLR 2 protège des souriceaux nouveau-nés de l’infection virale, ce qui tend à démontrer que la létalité due à l’infection par HSV-1 est liée à une réponse inflammatoire TLR 2-dépendante exagérée.
Protéines impliquées dans la réponse innée antivirale.
TLR 3
La découverte de TLR 3 et de son ligand, l’ARN bicaténaire (modélisé par son analogue synthétique, le poly I:C [14]) a, pour la première fois, suggéré que les TLR, en plus de leur rôle établi dans la défense antibactérienne, pouvaient jouer un rôle dans l’immunité antivirale. La caractérisation de TLR 3 a été suivie de nombreuses études mettant à profit la possibilité d’infecter des animaux déficients en TLR 3 (ou des cellules isolées de ces souris) par différents types de virus (Tableau I). Par ailleurs, des animaux présentant une déficience dans le gène codant pour l’adaptateur TRIF, essentiel à la production d’interféron de type I via TLR 3, présentent également une susceptibilité accrue après infection par le cytomégalovirus murin (MCMV) [15]. Ces travaux, qui ont permis de confirmer le rôle antiviral de la voie activée par TLR 3, ont néanmoins été remis en cause dans une étude récente au cours de laquelle des souris invalidées pour le gène Tlr 3 (TLR3-KO) ont été infectées par différents virus à des doses plus physiologiques [16]. De façon intéressante, il a également été montré que les souris TLR3-KO sont plus résistantes que les animaux contrôles à une infection létale par le virus du Nil occidental (WNV). Les auteurs ont observé, comme dans le cas décrit plus haut (TLR 2/HSV-1), une inflammation et une neuropathologie réduites chez les animaux déficients en TLR 3.
TLR 4
Identifié initialement comme composante du complexe liant le lipopolysaccharide (LPS), TLR 4 s’est vu, par la suite, attribuer des fonctions antivirales. Il a été montré que des animaux déficients en TLR 4 présentent un taux de réplication virale plus important après infection in vivo par le virus respiratoire syncytial (RSV). Ces travaux, confirmés ultérieurement par d’autres groupes, sont néanmoins l’objet de controverses [17], même si la capacité de TLR 4 à être activé en présence de protéines d’origine virale semble acquise. D’autres protéines virales, comme la protéine d’enveloppe du virus de la tumeur mammaire de la souris (MMTV), ou la glycoprotéine G du VSV, peuvent activer des cellules cibles de façon dépendante de TLR 4.
TLR 7/8
La nature de certains ligands synthétiques, comme des dérivés imidazoquinoliniques (imiquimod et resiquimod) ou des analogues de guanosine (loxoribine), capables d’activer les TLR 7/8, a permis d’identifier l’ARN simple brin comme le ligand naturel de ces récepteurs. En effet, ces molécules présentent des similitudes structurales avec l’ARN monocaténaire, ce qui a conduit à suggérer un rôle pour les TLR 7/8 dans la détection de certaines infections virales. Cette hypothèse s’est révélée exacte dans un certain nombre de cas (Tableau I).
TLR 9
Ce récepteur est capable de détecter l’ADN double brin non méthylé au niveau des séquences CpG, [18] et ce de façon séquence-spécifique [19]. La démonstration la plus convaincante du rôle essentiel de TLR 9 dans la lutte antivirale a été apportée par des études de la susceptibilité d’animaux mutants ou invalidés pour Tlr 9 après des infections in vivo par le MCMV. Cet agent infectieux, de la famille des β herpes virus, possède un génome formé d’une longue molécule d’ADN double brin et se prête donc idéalement à ces investigations. Trois études indépendantes ont montré que des animaux déficients pour TLR 9 présentent une hypersusceptibilité au MCMV, qui se manifeste par une réplication virale incontrôlée accompagnée d’une létalité accrue.
De l’ensemble de ces études, il apparaît clairement que la signalisation déclenchée par les récepteurs de la famille Toll constitue un élément essentiel de la lutte antivirale. Bien que les mécanismes sous-jacents ne soient pas totalement élucidés, le défaut de production d’interférons de type I chez les animaux TLR-déficients semble être la cause majeure pouvant expliquer leur hypersusceptibilité à ces infections. Par conséquent, les TLR constituent des cibles thérapeutiques de choix pour de nouvelles approches pharmacologiques des traitements antiviraux. L’utilisation, chez l’homme, d’agonistes de TLR 7 (comme l’isatoribine) pour le traitement d’infection par le virus de l’hépatite C (HCV) [20], ou dans les cas de verrues provoquées par le virus de papillome (avec l’imiquimod [21]), en est la démonstration.
L’analyse du Tableau I appelle également plusieurs commentaires : certains virus, comme le MCMV, paraissent capables d’activer plusieurs TLR (TLR 3 et 9, éventuellement TLR 2). Cette observation semble avoir une signification fonctionnelle, illustrée par la plus grande sensibilité au MCMV d’animaux déficients pour la protéine MyD88, utilisée comme adaptateur pour la transduction du signal en aval de la plupart des TLR (sauf TLR 3), par rapport à des souris mutantes pour TLR 9. Cela tendrait à prouver l’existence d’une synergie entre les TLR activés au cours d’une infection virale, synergie qui permettrait une protection maximale. Cette collaboration pourrait se manifester par une activation de types cellulaires différents (en fonction des TLR exprimés par ces cellules) ou par une activation des différents TLR échelonnée au cours du temps, en fonction de la progression de l’infection virale (les TLR exprimés en surface répondant plus précocement que les TLR capables de détecter des composants viraux intracellulaires).
Le rôle des TLR dans la défense antivirale chez l’homme est plus controversé. Des mécanismes de redondance entre les différents TLR entre eux, ou avec d’autres facteurs, ont été proposés afin d’expliquer l’absence d’hypersusceptibilité aux infections virales des patients déficients en IRAK 4, qui présentent un défaut de signalisation par les TLR 7, 8 et 9 [22]. Enfin, la variabilité des ligands reconnus par les TLR pose la question de la spécificité de leur reconnaissance : en effet, les TLR constituent une famille relativement homogène (Figure 1A), formée de protéines à domaine riche en leucine (LRR) dans leur partie extracellulaire et d’un domaine transmembranaire séparant la région TIR. La détermination de la structure des différents TLR associés à leurs ligands devrait apporter des réponses à cette question et permettre la modélisation d’agonistes ou d’antagonistes spécifiques de chaque interaction ligand-TLR. Enfin, les nombreux travaux portant sur la diversité des TLR chez l’homme [23] indiquent que l’étude de leur variabilité génétique dans la susceptibilité à diverses infections, notamment virales, constitue un sujet majeur de recherche dans les années à venir.
Réponse antivirale indépendante des TLR
La stimulation des TLR n’explique pas toutes les réponses antivirales innées. En effet, une induction des molécules de costimulation CD80 et CD86 a été observée en réponse au poly I:C chez des double mutants MyD88−/−/Trif−/−, pour qui toute stimulation via les TLR est virtuellement abolie. De plus, la localisation subcellulaire des TLR 3, 7 et 9 au niveau des endosomes rend improbable la détection de certains intermédiaires de réplication virale, comme l’ARN double brin, localisés au niveau cytoplasmique. Ces données ont permis de suggérer l’existence de détecteurs (« senseurs ») intracellulaires d’ARN double brin dont l’activation, indépendante des TLR, aboutit également à la production d’interférons de type I (Tableau I).
RIG-I (retinoic acid inducible gene)
Par approche fonctionnelle, via la recherche d’ADNc capables d’activer un gène rapporteur placé sous le contrôle d’un promoteur inductible par les interférons de type I, Yoneyama et ses collaborateurs ont identifié la protéine RIG-I [8]. Ce facteur (Figure 1B), qui contient un domaine hélicase ATP-dépendant ainsi que deux régions CARD, est capable de lier in vitro le poly I:C. La surexpression de RIG-I permet aux cellules infectées par le VSV de résister à des doses plus élevées de virus. Ces résultats ont été confirmés en utilisant des fibroblastes isolés de souris dans lesquelles le gène RIG-I était inactivé [7] : ces cellules deviennent plus susceptibles à une infection par le VSV et ne produisent pas d’interférons de type I après addition de virus Sendaï (SeV). L’étude de la voie de signalisation montre que les kinases TBK-1 et IKK-i sont impliquées, alors que l’effet dominant de la surexpression de RIG-I n’est pas bloqué par l’inactivation de la signalisation induite par les TLR.
MDA5 (melanoma differentiation-associated gene 5)
La recherche, dans les banques de données, de protéines possédant un domaine hélicase similaire à celui de RIG-I a mené à l’étude de MDA5 (Figure 1B). La surexpression de cette protéine en culture cellulaire, après activation de IRF-3 (un facteur essentiel pour la transcription des gènes codant pour les interférons de type I) et de la voie NF-κB, protège les cellules d’une infection virale. Un autre facteur à domaine hélicase, LPG2, présente une activité « dominant-négatif » en transfection cellulaire, et peut donc réguler négativement la voie RIG-I.
IPS-1 (interferon-β promoter stimulator 1)
Découverte simultanément par quatre groupes différents [9–12], IPS-1 (également nommée MAVS, VISA ou Cardif), qui ne contient qu’un domaine de type CARD (domaine d’interaction protéine-protéine, Figure 1B), induit, lors d’une surexpression, une réponse antivirale liée à l’activation des voies NF-κB et IRF. Ces propriétés semblent être dues à la capacité d’IPS-1 de s’associer aux protéines RIG-I et MDA5. Cependant, il est possible qu’IPS-1 agisse également sur l’induction de la production d’interféron par la voie TLR 3-dépendante, en s’associant avec TRAF6 [11]. Enfin, la localisation mitochondriale de cette protéine permet de suggérer un lien entre immunité antivirale et apoptose [10].
Plutôt que remplir le rôle de détecteur d’ARN bicaténaire, la protéine IPS-1 semble essentielle à l’induction des gènes codant pour les interférons en réponse à une stimulation par de l’ADN double brin sous forme B (conformation d’ADN physiologiquement la plus commune), et non sous forme Z [24]. Ces données, qui offrent de nouvelles possibilités dans le traitement des infections virales, répondent de surcroît à certaines questions jusque là inexpliquées. En effet, la capacité des souris Tlr9 −/− à produire une réponse immunitaire en réponse à une vaccination par l’ADN double brin pourrait trouver son origine dans l’existence d’un récepteur intracellulaire (qui pourrait être IPS-1) pour ce type de molécule.
PKR (protein kinase dsRNA-dependent)
Soupçonnée très tôt d’être un détecteur d’ARN bicaténaire, la PKR a effectivement été impliquée comme un facteur essentiel de la réponse antivirale (Tableau I). Cependant, des infections virales d’animaux chez lesquels la PKR et le complexe RNaseL/2’-5’ oligo-adénylate synthase (OAS) avait été inactivés ont montré l’existence de voies de résistance alternatives dépendantes de l’interféron.
LY49H
Ce récepteur, qui appartient à une famille de molécules exprimées à la surface des cellules NK chez la souris, n’est capable de détecter qu’un seul pathogène, le MCMV. L’interaction spécifique entre une protéine d’origine virale, homologue aux molécules de classe I d’histocompatibilité, et LY49H active les cellules NK par l’intermédiaire de l’adaptateur DAP12 [25], ce qui aboutit à la production d’interféron γ par ces cellules. Il est également apparu que l’activation des cellules NK par LY49H n’est pas suffisante pour conférer une protection efficace : les interférons de type I, produits par les macrophages ou les cellules dendritiques en réponse aux voies TLR ou RIG-I, mais également d’autres cytokines sont essentiels pour que les cellules NK puissent jouer leur rôle. L’importance de cette collaboration cellulaire est illustrée par les interactions entre cellules dendritiques et cellules NK, cruciales pour l’induction d’une réponse énergique contre l’infection par le MCMV [26, 27].
Mis à part LY49H et la PKR, pour lesquels des modèles animaux permettent de tester, in vivo, l’influence de l’absence de ces gènes sur la réponse innée, les facteurs de la réponse antivirale TLR-indépendante souffrent encore d’une description insuffisante. Dans le cas de RIG-I, l’effet délétère de l’invalidation (quelques animaux seulement survivent 3 semaines après leur naissance) empêche toute étude de l’infection par différents types de virus. Même si les données récoltées jusqu’ici présentent certaines incertitudes et contradictions (si IPS-1 est réellement un récepteur pour l’ADNdb, comment expliquer son rôle protecteur vis-à-vis d’une infection par le VSV ? Pourquoi et dans quel but peut-il interagir avec RIG-I et MDA5 ?), il est clair que cette nouvelle voie de réponse aux infections virales présente un intérêt considérable, tant d’un point de vue fondamental que thérapeutique.
Conclusions
Afin de synthétiser la participation de ces différents facteurs et pour rendre compte des phénomènes de redondance entre les signalisations TLR-dépendante et -indépendante, nous proposons un schéma, simplifié, dans lequel les acteurs de ces voies de signalisation seraient localisés dans des types cellulaires différents (Figure 2). Ces différentes voies convergent vers des mécanismes effecteurs qui culminent avec la production d’interférons, indispensables à l’établissement d’une immunité antivirale efficace. Les points de convergence sont vraisemblablement des adaptateurs de la famille TRAF et, en aval, les kinases TBK1 et IKK-i, responsables de la phosphorylation et de l’activation de facteurs de transcription (IRF 3, 5 et 7) indispensables à la biosynthèse d’interférons. La découverte de composés capables de stimuler l’activité de ces kinases, qui sont au centre de toutes les réponses antivirales, pourrait donc constituer une avancée thérapeutique considérable.

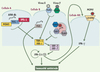 |
Figure 2. Voies de signalisation conduisant à une réponse innée antivirale. Dans ce modèle, différents types cellulaires (A ou B) sont capables de détecter une infection virale en fonction de la nature des récepteurs qu’elles expriment. Ces différentes voies convergent vers des kinases (TBK-1, IKK-i) qui permettent l’activation de facteurs de transcription (IRF 3 et 7) indispensables à la production d’interférons de type I (α/β). Ces cytokines pourront alors exercer des fonctions antivirales directes, ou indirectes par l’activation des cellules NK, activées en réponse au cytomégalovirus murin (MCMV) se liant au récepteur LY49H. Ce schéma n’exclut pas la possibilité (non représentée ici pour des raisons de simplicité) que différents détecteurs puissent être exprimés par un même type cellulaire, comme c’est le cas pour les cellules NK qui expriment à la fois LY49H et TLR 3. |
Remerciements
Les travaux de notre laboratoire bénéficient du soutien financier de la Fondation pour la recherche médicale (FRM), de la Ligue contre le cancer, de l’Association pour la recherche contre le cancer (ARC), de l’Agence de biomédecine et de l’Université Louis-Pasteur.
Article reçu le 7 décembre 2005, accepté le 1er février 2006.
Références
- Hoebe K, Janssen E, Beutler B. The interface between innate and adaptive immunity. Nat Immunol 2004; 5 : 971–4. [Google Scholar]
- Hoffmann JA. The immune response of Drosophila. Nature 2003; 426 : 33–8. [Google Scholar]
- Flajnik MF, Du Pasquier L. Evolution of innate and adaptive immunity : can we draw a line ? Trends Immunol 2004; 25 : 640–4. [Google Scholar]
- Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol 2005; 17 : 1–14. [Google Scholar]
- Beutler B. Inferences, questions and possibilities in Toll-like receptor signalling. Nature 2004; 430 : 257–63. [Google Scholar]
- Bowie AG, Haga IR. The role of Toll-like receptors in the host response to viruses. Mol Immunol 2005; 42 : 859–67. [Google Scholar]
- Kato H, Sato S, Yoneyama M, et al. Cell type-specific involvement of RIG-I in antiviral response. Immunity 2005; 23 : 19–28. [Google Scholar]
- Yoneyama M, Kikuchi M, Natsukawa T, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol 2004; 5 : 730–7. [Google Scholar]
- Kawai T, Takahashi K, Sato S, et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol 2005; 6 : 981–8. [Google Scholar]
- Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005; 122 : 669–82. [Google Scholar]
- Xu LG, Wang YY, Han KJ, et al. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell 2005; 19 : 727–40. [Google Scholar]
- Meylan E, Curran J, Hofmann K, et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005; 437 : 1167–72. [Google Scholar]
- Kurt-Jones EA, Chan M, Zhou S, et al. Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc Natl Acad Sci USA 2004; 101 : 1315–20. [Google Scholar]
- Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 2001; 413 : 732–8. [Google Scholar]
- Hoebe K, Du X, Georgel P, et al. Identification of Lps2 as a key transducer of MyD88-independent TIR signaling. Nature 2003; 424 : 743–8. [Google Scholar]
- Edelmann KH, Richardson-Burns S, Alexopoulou L, et al. Does Toll-like receptor 3 play a biological role in virus infections ? Virology 2004; 322 : 231–8. [Google Scholar]
- Ehl S, Bischoff R, Ostler T, et al. The role of Toll-like receptor 4 versus interleukin-12 in immunity to respiratory syncytial virus. Eur J Immunol 2004; 34 : 1146–53. [Google Scholar]
- Hemmi H, Takeuchi O, Kawai T, et al. A Toll-like receptor recognizes bacterial DNA. Nature 2000; 408 : 740–5. [Google Scholar]
- Bauer S, Kirschning CJ, Hacker H, et al. Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc Natl Acad Sci USA 2001; 98 : 9237–42. [Google Scholar]
- Horsmans Y, Berg T, Desager JP, et al. Isatoribine, an agonist of TLR7, reduces plasma virus concentration in chronic hepatitis C infection. Hepatology 2005; 42 : 724–31. [Google Scholar]
- Jacobs S, Grussendorf-Conen EI, Rosener I, Rubben A. Molecular analysis of the effect of topical imiquimod treatment of HPV 2/27/57-induced common warts. Skin Pharmacol Physiol 2004; 17 : 258–66. [Google Scholar]
- Yang K, Puel A, Zhang S, et al. Human TLR-7-, -8-, and -9-mediated induction of IFN-alpha/beta and -lambda is IRAK-4 dependent and redundant for protective immunity to viruses. Immunity 2005; 23 : 465–78. [Google Scholar]
- Schroder NW, Schumann RR. Single nucleotide polymorphisms of Toll-like receptors and susceptibility to infectious disease. Lancet Infect Dis 2005; 5 : 156–64. [Google Scholar]
- Ishii KJ, Coban C, Kato H, et al. A Toll-like receptor-independent antiviral response induced by double-stranded B-form DNA. Nat Immunol 2006; 7 : 40–8. [Google Scholar]
- Sjolin H, Tomasello E, Mousavi-Jazi M, et al. Pivotal role of KARAP/DAP12 adaptor molecule in the natural killer cell-mediated resistance to murine cytomegalovirus infection. J Exp Med 2002; 195 : 825–34. [Google Scholar]
- Andoniou CE, van Dommelen SL, Voigt V, et al. Interaction between conventional dendritic cells and natural killer cells is integral to the activation of effective antiviral immunity. Nat Immunol 2005; 6 : 1011–9. [Google Scholar]
- Andrews DM, Scalzo AA, Yokoyama WM, et al. Functional interactions between dendritic cells and NK cells during viral infection. Nat Immunol 2003; 4 : 175–81. [Google Scholar]
- Bieback K, Lien E, Klagge IM, et al. Hemagglutinin protein of wild-type measles virus activates toll-like receptor 2 signaling. J Virol 2002; 76 : 8729–36. [Google Scholar]
- Compton T, Kurt-Jones EA, Boehme KW, et al. Human cytomegalovirus activates inflammatory cytokine responses via CD14 and Toll-like receptor 2. J Virol 2003; 77 : 4588–96. [Google Scholar]
- Zhou S, Kurt-Jones EA, Mandell L, et al. MyD88 is critical for the development of innate and adaptive immunity during acute lymphocytic choriomeningitis virus infection. Eur J Immunol 2005; 35 : 822–30. [Google Scholar]
- Wang JP, Kurt-Jones EA, Shin OS, et al. Varicella-zoster virus activates inflammatory cytokines in human monocytes and macrophages via Toll-like receptor 2. J Virol 2005; 79 : 12658–66. [Google Scholar]
- Duesberg U, von dem BA, Kirschning C, et al. Cell activation by synthetic lipopeptides of the hepatitis C virus (HCV)-core protein is mediated by Toll like receptors (TLRs) 2 and 4. Immunol Lett 2002; 84 : 89–95. [Google Scholar]
- Wang T, Town T, Alexopoulou L, et al. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med 2004; 10 : 1366–73. [Google Scholar]
- Hewson CA, Jardine A, Edwards MR, et al. Toll-like receptor 3 is induced by and mediates antiviral activity against rhinovirus infection of human bronchial epithelial cells. J Virol 2005; 79 : 12273–9. [Google Scholar]
- Ashkar AA, Yao XD, Gill N, et al. Toll-like receptor (TLR)-3, but not TLR4, agonist protects against genital herpes infection in the absence of inflammation seen with CpG DNA. J Infect Dis 2004; 190 : 1841–9. [Google Scholar]
- Guillot L, Le GR, Bloch S, et al. Involvement of Toll-like receptor 3 in the immune response of lung epithelial cells to double-stranded RNA and influenza A virus. J Biol Chem 2005; 280 : 5571–80. [Google Scholar]
- Tabeta K, Georgel P, Janssen E, et al. Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc Natl Acad Sci USA 2004; 101 : 3516–21. [Google Scholar]
- Kurt-Jones EA, Popova L, Kwinn L, et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat Immunol 2000; 1 : 398–401. [Google Scholar]
- Yang R, Murillo FM, Delannoy MJ, et al. B lymphocyte activation by human papillomavirus-like particles directly induces Ig class switch recombination via TLR4-MyD88. J Immunol 2005; 174 : 7912–9. [Google Scholar]
- Triantafilou K, Triantafilou M. Coxsackievirus B4-induced cytokine production in pancreatic cells is mediated through toll-like receptor 4. J Virol 2004; 78 : 11313–20 [Google Scholar]
- Jiang Z, Georgel P, Du X, et al. CD14 is required for MyD88-independent LPS signaling. Nat Immunol 2005; 6 : 565–70. [Google Scholar]
- Rassa JC, Meyers JL, Zhang Y, et al. Murine retroviruses activate B cells via interaction with toll-like receptor 4. Proc Natl Acad Sci USA 2002; 99 : 2281–6. [Google Scholar]
- Lund JM, Alexopoulou L, Sato A, et al. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc Natl Acad Sci USA 2004, 101 : 5598–603. [Google Scholar]
- Diebold SS, Kaisho T, Hemmi H, et al. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 2004, 303 : 1529–31. [Google Scholar]
- Krug A, Luker GD, Barchet W, et al. Herpes simplex virus type 1 activates murine natural interferon-producing cells through toll-like receptor 9. Blood 2004; 103 : 1433–7. [Google Scholar]
- Lund J, Sato A, Akira S, et al. Toll-like receptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J Exp Med 2003; 198 : 513–20. [Google Scholar]
- Krug A, French AR, Barchet W, et al. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity 2004; 21 : 107–19. [Google Scholar]
- Delale T, Paquin A, Asselin-Paturel C, et al. MyD88-dependent and -independent murine cytomegalovirus sensing for IFN-alpha release and initiation of immune responses in vivo. J Immunol 2005; 175 : 6723–32. [Google Scholar]
- Yoneyama M, Kikuchi M, Matsumoto K, et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol 2005; 175 : 2851–8. [Google Scholar]
- Balachandran S, Roberts PC, Brown LE, et al. Essential role for the dsRNA-dependent protein kinase PKR in innate immunity to viral infection. Immunity 2000; 13 : 129–41. [Google Scholar]
- Flodstrom-Tullberg M, Hultcrantz M, Stotland A, et al. RNase L and double-stranded RNA-dependent protein kinase exert complementary roles in islet cell defense during coxsackievirus infection. J Immunol 2005; 174 : 1171–7. [Google Scholar]
- Daniels KA, Devora G, Lai WC, et al. Murine cytomegalovirus is regulated by a discrete subset of natural killer cells reactive with monoclonal antibody to Ly49H. J Exp Med 2001; 194 : 29–44. [Google Scholar]
- Arase H, Mocarski ES, Campbell AE, et al. Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science 2002; 296 : 1323–6. [Google Scholar]
- Smith HR, Heusel JW, Mehta IK, et al. Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc Natl Acad Sci USA 2002; 99 : 8826–31. [Google Scholar]
- Lee SH, Zafer A, de RY, et al. Transgenic expression of the activating natural killer receptor Ly49H confers resistance to cytomegalovirus in genetically susceptible mice. J Exp Med 2003; 197 : 515–26. [Google Scholar]
Liste des tableaux
Liste des figures
|
Figure 1. Molécules impliquées dans la réponse antivirale TLR-dépendante (A) et TLR-indépendante (B). Différents domaines, identifiés par SMART (simple modular architecture research tool), sont représentés. LRR : régions riches en leucine ; TIR : Toll-interleukin 1 receptor ; TM : domaine transmembranaire ; CARD : domaine de recrutement des caspases ; DEXDc : domaine dead des protéines à hélicase ; DSRM : domaine de liaison à l’ARN double brin ; Lect-C : lectine de type C. La localisation cellulaire de ces protéines est indiquée (RE : réticulum endoplasmique). |
| Dans le texte | |
|
Figure 2. Voies de signalisation conduisant à une réponse innée antivirale. Dans ce modèle, différents types cellulaires (A ou B) sont capables de détecter une infection virale en fonction de la nature des récepteurs qu’elles expriment. Ces différentes voies convergent vers des kinases (TBK-1, IKK-i) qui permettent l’activation de facteurs de transcription (IRF 3 et 7) indispensables à la production d’interférons de type I (α/β). Ces cytokines pourront alors exercer des fonctions antivirales directes, ou indirectes par l’activation des cellules NK, activées en réponse au cytomégalovirus murin (MCMV) se liant au récepteur LY49H. Ce schéma n’exclut pas la possibilité (non représentée ici pour des raisons de simplicité) que différents détecteurs puissent être exprimés par un même type cellulaire, comme c’est le cas pour les cellules NK qui expriment à la fois LY49H et TLR 3. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.