")
")
| Issue |
Med Sci (Paris)
Volume 22, Number 10, Octobre 2006
|
|
|---|---|---|
| Page(s) | 837 - 844 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/20062210837 | |
| Published online | 15 octobre 2006 | |
Microscopie à deux photons pour l’imagerie cellulaire fonctionnelle : avantages et enjeux ou Un photon c’est bien… mais deux c’est mieux !
Two-photon laser scanning fluorescence microscopy for functional cellular imaging: advantages and challenges or One photon is good… but two is better!
1
Centre d’optique, photonique et laser, Département de physique, de génie physique et d’optique, Université Laval, Québec, G1K 7P4 Canada
2
Département de Psychiatrie, Université Laval, Unité de Neurobiologie Cellulaire, Centre de recherché Université Laval Robert-Giffard, 2601, chemin de la Canardière, Québec, G1J 2G3 Canada
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
L’observation de la dynamique des événements moléculaires dans la cellule in situ présente une série de défis, notamment la capacité de suivre ces événements avec le maximum de résolution spatiale et temporelle tout en minimisant l’interférence avec la biologie du tissu et de la cellule. L’exploitation récente d’approches fondées sur l’optique non-linéaire, telle que la microscopie par balayage laser de fluorescence produite par excitation à deux photons, a permis de faire des progrès énormes dans ce domaine, notamment parce qu’elle permet de faire des mesures dans un espace très confiné à l’intérieur du tissu intact et à des profondeurs inaccessibles avec la microscopie linéaire conventionnelle. En minimisant l’excitation indésirable du tissu en dehors du point focal, on améliore la résolution et la sensibilité, on simplifie le système optique et on minimise la phototoxicité. Ces avantages sont à la source du succès de la microscopie à deux photons pour l’imagerie cellulaire fonctionnelle. Des percées récentes en optique/photonique permettent d’envisager d’améliorer davantage la résolution spatiale et temporelle de ce type d’imagerie et la capacité de sonder encore plus profondément dans le tissu pour repousser les limites de la biochimie fonctionnelle et de la biologie cellulaire actuelles.
Abstract
One of the main challenges of modern biochemistry and cell biology is to be able to observe molecular dynamics in their functional context, i.e. in live cells in situ. Thus, being able to track ongoing molecular events with maximal spatial and temporal resolution (within subcellular compartments), while minimizing interference with tissue biology, is key to future developments for in situ imaging. The recent use of non-linear optics approaches in tissue microscopy, made possible in large part by the availability of femtosecond pulse lasers, has allowed major advances on this front that would not have been possible with conventional linear microscopy techniques. Of these approaches, the one that has generated most advances to date is two-photon laser scanning fluorescence microscopy. While this approach does not really provide improved resolution over linear microscopy in non absorbing media, it allows us to exploit a window of low absorbance in live tissue in the near infrared range. The end result is much improved tissue penetration, minimizing unwanted excitation outside the focal area, which yields an effective improvement in resolution and sensitivity. The optical system is also simplified and, more importantly, phototoxicity is reduced. These advantages are at the source of the success of two-photon microscopy for functional cellular imaging in situ. Yet, we still face further challenges, reaching the limits of resolution that conventional optics can offer. Here we review some recent advances in optics/photonics approaches that hold promises to improve our ability to probe the tissue in finer areas, at faster speed, and deeper into the tissue. These include super-resolution techniques, introduction of non paraxial optics in microscopy and use of amplified femtosecond lasers, yielding enhanced spatial and temporal resolution as well as tissue penetration.
© 2006 médecine/sciences - Inserm / SRMS
Un des enjeux majeurs de la biochimie et de la biologie cellulaire actuelle est de pouvoir suivre de manière dynamique les événements moléculaires à l’intérieur de la cellule vivante dans son contexte fonctionnel, c’est-à-dire in situ (dans son tissu d’origine). Il apparaît de plus en plus évident que des facteurs spatio-temporels très fins à l’intérieur de la cellule vivante déterminent en grande partie la spécificité des signaux cellulaires. Il suffit de penser aux fluctuations calciques intracellulaires qui participent à une multitude de cascades de signalisation ; sans spécificité spatiale et temporelle, la cellule ne pourrait utiliser les signaux calciques de manière utile et efficace.
Parmi les techniques d’imagerie non-linéaire modernes, la microscopie par excitation à deux photons (M2P) est celle qui a permis de faire les plus grands progrès en ce sens, permettant d’étudier à haute résolution non seulement la structure, mais aussi le fonctionnement moléculaire dans le temps, à l’intérieur de la cellule. Nous passerons ici en revue quelques principes qui sont à la base des avantages de cette technique afin de mettre en relief certains des défis qui demeurent (résolutions, sensibilité, profondeur de pénétration dans le tissu et phototoxicité) pour l’imagerie cellulaire fonctionnelle. Les exemples présentés sont tirés des neurosciences puisque l’étude du neurone présente probablement les défis les plus grands en termes de résolution spatiale et temporelle. En effet, la taille de certaines ramifications et spécialisations neuronales (« 1 µm) et la rapidité des signaux neurochimiques, comme ceux qui sont impliqués dans la transmission synaptique (de l’ordre de la ms), sont à la limite de ce que la microscopie optique actuelle permet d’étudier.
La microscopie à deux photons : principes de base
Il est possible pour un atome ou une molécule d’absorber « simultanément » deux photons (2P) ou plus (Figure 1A). On appelle ce phénomène l’absorption à deux photons (A2P). Il a été prédit par Göper-Mayer en 1931 [1] et observé 30 ans plus tard par Kaiser et Garrett [2]. La première observation d’A2P dans un milieu organique a été effectuée par Peticolas et al. [3] quelques années plus tard et la microscopie laser fondée sur l’A2P a été proposée par Sheppard et Kompfner [4]. Cependant, c’est seulement en 1990 que Denk et al. [5] ont rapporté la première application expérimentale de l’A2P à la microscopie laser. Ce type de microscopie est survenu beaucoup plus tard que la découverte de l’A2P puisqu’elle nécessitait l’utilisation d’un laser ayant des impulsions sub-picoseconde. En effet, l’intensité lumineuse utilisée doit être très grande pour que la probabilité soit non négligeable que deux photons passent près de l’atome à exciter dans un laps de temps suffisamment court (10-16 s) pour que leurs actions s’additionnent. Des impulsions de très courte durée (< 200 fs) sont nécessaires afin d’atteindre des intensités suffisantes à l’observation de ce phénomène (voir plus loin).
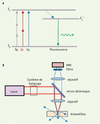 |
Figure 1. Principes de microscopie multiphotonique. A. Schématisation des niveaux d’énergie d’une molécule fluorescente. La flèche bleue représente l’absorption à un photon (1p), les flèches rouges représentent l’absorption à 2 photons (2p) et les grises représentent l’absorption à 3 photons (3p). La flèche pointillée représente le passage vers un niveau d’énergie intermédiaire avant l’émission spontanée d’un photon de fluorescence (flèche verte). B. Schéma expérimental du montage de la M2P conventionnelle. La source d’excitation est dirigée vers l’échantillon via un système de balayage et un miroir dichroïque, puis elle est concentrée au point focal d’excitation par l’objectif. La fluorescence émise par l’échantillon est ensuite collectée par le même objectif, filtrée par un filtre d’émission et détectée par un tube photomultiplicateur (PMT). Le volume d’excitation doit être éclairé pendant un certain temps (appelé pixel dwell time) afin que l’on puisse récolter assez de photons de fluorescence pour produire un signal clair. |
En M2P, afin d’exciter efficacement les molécules fluorescentes, on focalise les impulsions laser dans un tout petit volume (Figure 1B). Dans ce volume, les molécules de l’échantillon absorbent 2P ayant chacun la moitié de l’énergie (donc le double de la longueur d’onde) nécessaire à leur excitation ; celles-ci réémettent alors généralement un photon (1P) par fluorescence. On peut ensuite reconstituer l’image d’un plan xy de l’échantillon en déplaçant le point focal dans ce plan [6]. On obtient des images en trois dimensions en balayant plusieurs plans à des profondeurs différentes.
Résolution spatiale
La Figure 2A compare l’intensité (absorption à 1P) et l’intensité au carré (2P) de taches focales pour un même objectif, à une longueur d’onde de 450 nm et 800 nm. L’absorption par les molécules fluorescentes étant proportionnelle à l’intensité au carré dans le cas de la M2P [7, 8], cela améliore la résolution pour une longueur d’onde donnée. Le résultat net est que, théoriquement, la résolution radiale est légèrement inférieure avec une excitation à 2P à 800 nm qu’avec 1P à 450 nm (Figure 2A). En revanche, l’intensité au carré diminue plus rapidement sur l’axe z dans le cas à 2P. Cette illustration ne considère cependant pas l’absorption et la diffusion dans les tissus. Or, le coefficient d’absorption de plusieurs des composantes des tissus est au moins 10 fois moins important à 800 qu’à 450 nm (Figure 2B). La diffusion est également inférieure à une longueur d’onde de 800 nm. En microscopie confocale (à 1P), il y a donc beaucoup plus de fluorescence émise hors du point focal (Figure 2C). Cette fluorescence hors foyer devient une source de bruit importante, minant la résolution et la sensibilité du système ; de plus, elle affecte le tissu (phototoxicité) hors du point d’investigation. Pour maintenir une bonne résolution en microscopie confocale, on utilise un sténopé, afin de rejeter les photons émis par fluorescence hors du point focal. Pour un tissu épais (>100 µm), on doit excessivement réduire le diamètre du sténopé et augmenter l’intensité de la lumière incidente. Ceci rend rapidement l’intensité lumineuse hors foyer trop importante, compromettant la détection du signal et l’intégrité du tissu.
 |
Figure 2. Avantages et limites de la M2P pour la résolution spatiale. A. Images 2D du profil d’intensité lumineuse de points focaux (1P ; dans le plan xz ou yz à 450 et 800 nm, respectivement) et intensité au carré à 800 nm (2P) obtenus par simulation. L’ouverture numérique de l’objectif est de 0,5 et le coefficient d’absorption est nul. La perte de résolution en utilisant une longueur d’onde plus grande (la résolution est proportionnelle à la largeur à mi-hauteur des points focaux) se trouve compensée par le fait que l’absorption de 2P est proportionnelle à l’intensité au carré. Le résultat net est que la résolution radiale avec une excitation à 2P à 800 nm est comparable à celle obtenue avec 1P à 450 nm. B. Coefficient d’absorption des principaux absorbeurs dans les tissus en fonction de la longueur d’onde. Notez l’absorption 10 à 100 fois plus faible à 800 nm qu’à 450 nm (flèches) due à la présence d’éléments absorbant dans le tissu (mélamine, hémoglobine…). C. Profil de l’intensité lumineuse à 1P (gauche) et profil de l’intensité au carré à 2P (droite) obtenus par simulation en tenant compte, cette fois-ci, du coefficient d’absorption. La valeur du coefficient d’absorption est de 85 cm-1 dans le cas à 1P et de 8,5 cm-1 dans le cas à 2P (ouverture numérique = 0,16). D. Image d’un neurone pyramidal du néocortex exprimant la green fluorescent protein (GFP) à faible grossissement à gauche et avec un grossissement croissant vers la droite pour illustrer le détail des prolongements du neurone : le panneau du milieu met en évidence une dendrite ; le panneau de droite les détails d’épines dendritiques. Comme les épines ont une taille de l’ordre du micromètre, la résolution optique standard ne permet pas de distinguer leur structure fine. E. Exemple de technique utilisée pour augmenter la résolution en deçà des limites de diffraction de la lumière : la STED (stimulated emission depletion). On excite l’échantillon avec une impulsion laser de la même façon qu’on le fait en M2P. Par la suite, avant que les fluorophores réémettent la fluorescence, on désexcite les molécules autour du point focal par émission stimulée avec une impulsion en forme de beigne. Le volume d’excitation correspond alors à une soustraction entre le volume d’excitation et le volume de désexcitation autour du point focal. Les molécules encore excitées sont alors dans un volume beaucoup plus petit que le point d’excitation engendré par l’excitation, ce qui améliore la résolution radiale. (E1) présente le profil d’intensité de l’impulsion laser d’excitation, (E2) le profil d’intensité du faisceau en beigne qui suit l’impulsion d’excitation. (E3) montre le profil d’excitation résultant après le passage des deux premières impulsions, (E4) le profil inverse du beigne. |
Ainsi, considérant à la fois le diamètre fini du sténopé, l’importante absorption et la diffusion dans les tissus biologiques aux courtes longueurs d’onde, la M2P permet l’acquisition d’images à de plus grandes profondeurs, tout en ayant une meilleure résolution radiale et axiale en minimisant la taille de la région excitée et le stress imposé au tissu [9].
Il est possible d’utiliser l’absorption à trois photons pour augmenter la résolution spatiale ; celle-ci demeure cependant aussi limitée par la diffraction. Or, plusieurs mouvements moléculaires se font sur des échelles spatiales bien plus petites ; de même, plusieurs compartiments cellulaires (comme l’épine dendritique d’un neurone ; Figure 2D) sont eux-mêmes inférieurs en taille aux limites de diffraction de la lumière. Beaucoup d’efforts sont déployés pour obtenir des résolutions « 100 nm. Mentionnons, comme nouvelles approches, la microscopie 4Pi-confocale, la RESOLFT (reversible saturable optical fluorescence transition), l’illumination structurée et la STED (stimulated emission depletion) [10–13] (Figure 2E). Ces techniques sont cependant complexes et requièrent des alignements de laser très précis. En revanche, on assiste actuellement à l’émergence d’autres approches complémentaires moins complexes [14].
Maximisation du signal de fluorescence/imagerie en profondeur
La M2P offre une meilleure pénétration dans les tissus pour l’imagerie cellulaire par rapport à la microscopie confocale conventionnelle (Figure 3A). Celle-ci demeure toutefois limitée et son amélioration un enjeu important. À cette fin, il faut maximiser la fluorescence provenant du point focal et la détection de ces photons.
 |
Figure 3. Imagerie fonctionnelle en profondeur dans le tissu. A. Image reconstituée de cellules corticales dans le cerveau intact de souris transgéniques exprimant une protéine fluorescente dans une sous-population de neurones corticaux de la couche V. Une projection sur le côté de l’empilement des images permet de visualiser les cellules dans l’axe des z et d’illustrer la capacité d’imager en profondeur dans la matière grise du cortex cérébral (tirée de [16]). B. Graphique de l’intensité en fonction du temps pour un faisceau laser continu (noir) et pour des faisceaux laser pulsés à 100 MHz (bleu), à 10 MHz (vert) et à 250 kHz (rouge). Les faisceaux à 10 MHz et 250 kHz sont engendrés à l’aide d’un amplificateur régénératif. Notons que les quatre faisceaux laser ont la même puissance moyenne (correspondant à la puissance du faisceau continu). C. Imagerie fonctionnelle sur un bouton synaptique. (C1) Schéma de la circuiterie de l’hippocampe chez le rat. Les cellules granulaires (CG) sont situées dans le gyrus denté et envoient leur prolongement axonal dans la région CA3 où ils font synapse au niveau de dendrites proximales des cellules pyramidales. Les boutons synaptiques des CG peuvent être marqués à l’aide d’un indicateur calcique qui servira à rapporter les changements calciques de cette terminaison suite à une stimulation électrique des axones. (C2) Terminaisons en boutons marquées à l’aide de l’indicateur calcique Oregon Green BAPTA1-AM. La terminaison imagée est encadrée en pointillés. (C3) Pour maximiser la résolution temporelle on ne balaye qu’une seule ligne qui passe à travers le bouton. (C4) Les changements de fluorescence sont rapportés sous forme graphique dans le temps. D. Imagerie fonctionnelle d’un large réseau de neurones. (D1) Image en M2P de fluorescence provenant d’un marqueur calcique capturé par un grand nombre de neurones corticaux (plusieurs centaines de neurones peuvent être suivis avec cette méthode). (D2) Exemple de tracés rapportant la fluctuation de fluorescence dans le temps dans deux des neurones du champ en d1. (D3) Ces fluctuations peuvent être comptées à partir d’un seuil de détection donné et (D4) l’activité du réseau peut être reconstituée tel que sur ce raster-plot où chaque ligne représente un neurone (modifié à partir de [31]). |
Puissance, focalisation et impulsions courtes
Comme la M2P est un phénomène non-linéaire, maximiser l’intensité lumineuse a un impact particulièrement important sur le signal : en doublant l’intensité lumineuse d’excitation, la fluorescence émise est quadruplée. Cependant, une intensité moyenne lumineuse plus élevée (en photon cm-2 s-1) causera une augmentation du dommage au tissu. Il est donc préférable d’optimiser d’autres paramètres. On peut minimiser la taille du point focal en utilisant, par exemple, des objectifs avec la plus grande ouverture numérique possible : focaliser un faisceau laser d’une taille de 1 mm sur 1 mm fait gagner un facteur 106 en intensité crête.
En M2P, le fait de focaliser un faisceau laser intense en un tout petit volume n’est cependant pas suffisant pour obtenir assez de signal de fluorescence dans un court laps de temps (pixel dwell time). Il faut donc exploiter la possibilité de réduire la durée des impulsions laser pour en augmenter l’intensité. Comme la fluorescence dépend de la moyenne de l’intensité au carré [15], avec des impulsions de 100 fs et un taux de répétition de 80 MHz, gamme typique des lasers femtoseconde utilisés en microscopie, l’intensité lumineuse maximale est augmentée par un facteur 105 (donc la fluorescence, par un facteur 1010) sans changer l’intensité moyenne.
Taux de répétition du laser
Pour une impulsion laser d’une largeur temporelle τ et d’un taux de répétition fr, le signal de fluorescence est proportionnel à 1/(τfr)n-1 [16], où n est le nombre de photons absorbés (n = 2 pour l’A2P). Pour augmenter le signal, on peut donc envisager d’utiliser des impulsions plus courtes et un taux de répétition plus faible. Cependant, des impulsions de durée inférieure à 100 fs subiraient un élargissement temporel (dispersion) dans les composantes optiques et dans les tissus. D’autre part, pour réduire fr, l’utilisation d’un amplificateur régénératif permet d’additionner plusieurs impulsions successives afin de produire une impulsion géante (Figure 3B). L’intensité crête peut être multipliée par un facteur 108 en réduisant le taux de répétition à 1 Hz [17] sans changer l’intensité moyenne. En imagerie, puisqu’il faut au moins une impulsion par pixel, on réduit fr par un facteur plus petit ; pour un pixel dwell time de 4-10 μs, on doit réduire le taux de répétition à 100-250 kHz. Puisque cette technique augmente le signal de fluorescence, elle permet aussi d’imager plus en profondeur dans les échantillons [18]. L’apparition significative d’excitation à 2P hors du point focal semble toutefois limiter la profondeur d’imagerie autour de 1 mm [19].
Section efficace des fluorophores
Bien que certains fluorophores aient les mêmes caractéristiques pour l’absorption à 1P ou 2P, plusieurs ont un comportement différent. Certaines molécules comme le NADH (molécule endogène) ont une faible section efficace à 2P tandis que d’autres (GFP, CFP) ont une section efficace acceptable (~100 GM). Un des avantages notables des nanocristaux fluorescents de CdSe-ZnS, dits points quantiques (quantum dots), et qui fait leur popularité croissante en microscopie pour le suivi moléculaire [20], est qu’ils ont une section efficace près de 50 000 GM [21]. En M2P, les profils d’excitation sont également souvent plus large qu’en 1P, permettant d’exciter plus facilement deux fluorophores avec la même longueur d’onde, ce qui peut être très avantageux pour l’analyse par corrélation croisée de plusieurs signaux [22].
Approche alternative
La venue de micro-endoscopes utilisant la technologie du M2P permet d’aller beaucoup plus en profondeur in vivo, particulièrement en neurobiologie où les caractéristiques du tissu se prêtent bien à l’utilisation de cette approche [23] ; il s’agit néanmoins d’une technique plus invasive.
Résolution temporelle
L’imagerie fonctionnelle va au-delà de la morphologie, car elle fait intervenir une dimension supplémentaire, soit le temps. Elle permet donc de suivre l’évolution des composantes présentes dans un environnement vivant et d’étudier les événements et les fonctions reliées à ces composantes. Bien que l’imagerie fonctionnelle fasse souvent référence à des techniques telles que l’imagerie à résonance magnétique (IRM) ou l’imagerie par tomographie à émission de positons (PET), la résolution spatiale nettement supérieure qu’offre la M2P en fait une technique de choix pour l’imagerie fonctionnelle cellulaire et subcellulaire.
Pour disposer d’une bonne résolution temporelle, il faut optimiser le signal de fluorescence, optimiser la détection du signal, optimiser la vitesse de balayage et imager seulement le volume d’intérêt.
Compartimentalisation cellulaire
Le neurone se distingue par son activité électrique et une compartimentalisation de ses sous-unités fonctionnelles (dendrites, axones…). Les méthodes d’électrophysiologie modernes ont permis de faire de grandes avancées pour étudier les événements électriques dans les cellules nerveuses [24] mais ces méthodes se butent à certaines limites. L’une d’elles est la difficulté de suivre les courants synaptiques dans des compartiments très éloignés du cœur de la cellule (boutons synaptiques, ramifications dendritiques). L’imagerie cellulaire fonctionnelle permet de briser ces barrières spatiales et permet d’échantillonner les événements cellulaires dynamiques en plusieurs points d’un même neurone [25–27]. La rapidité de certains événements électriques neuronaux (typiquement de l’ordre de 1-100 ms) présente cependant des défis importants.
La méthode utilisée pour balayer le point focal à l’intérieur de l’échantillon détermine en partie les limites de résolution temporelle de la M2P. On peut déplacer l’objectif ou l’échantillon lui-même ; en pratique, on préfère balayer le faisceau laser sur la lentille (notamment avec des miroirs montés sur des galvanomètres). Avec cette configuration, il est possible d’obtenir une image 2D de 512x512 points en 1/30 s [28], ce qui fournit une résolution temporelle suffisante sur champ large pour suivre la dynamique d’un bon nombre de phénomènes biologiques. Pour certains signaux synaptiques, 30 images/s demeure cependant trop lent. Pour y remédier, on peut limiter l’échantillonnage à une aire réduite, voire à une seule ligne de balayage, comme pour l’imagerie fonctionnelle d’événements synaptiques (Figure 3C) [25–27]. Une autre approche prometteuse est l’utilisation de déflecteurs acousto-optiques, éléments non-mécaniques qui permettent donc d’envisager des déplacements de faisceau beaucoup plus rapides [29].
Activité du réseau
Un des enjeux pour comprendre l’activité neuronale se situe à un autre extrême, celui de suivre l’activité d’un vaste réseau de neurones à l’intérieur du tissu intact. Encore une fois, par rapport aux techniques d’électrophysiologie classique, l’imagerie cellulaire fonctionnelle à 2P offre une solution efficace pour échantillonner l’activité d’un grand nombre de neurones en profondeur dans le tissu (Figure 3D) [30–32]. Cela dit, la haute résolution axiale de la M2P oblige à prendre un grand nombre d’images dans l’axe des z pour capturer l’état d’objets dispersés dans un échantillon épais de tissu, ce qui limite d’autant la résolution temporelle des phénomènes dynamiques à observer. Pour l’étude d’un réseau de neurones dans le cerveau, par exemple, il faut pouvoir obtenir une image à grande vitesse de tout le volume de tissu pour détecter les décharges successives des cellules qui se produisent sur une échelle de temps de quelques ms.
Microscopie avec axicon
Plusieurs approches ont été envisagées pour accélérer l’acquisition d’image en M2P à balayage laser, notamment l’utilisation de matrices de lentilles [33]. Si les cellules à l’étude ne sont pas trop densément groupées, on peut éliminer le besoin de scanner le volume dans l’axe des z, puisque cette information n’est pas nécessairement utile. Pour cela, on peut envisager d’augmenter la profondeur de champ. L’enjeu est cependant de ne pas sacrifier la résolution radiale (dans le plan xy), ce qui se produit avec des objectifs classiques en diminuant l’ouverture numérique, par exemple.
Plusieurs techniques tentent d’augmenter la profondeur de champ des lentilles, comme l’utilisation d’axilens [34] ou de lentilles bi-réfringentes entre deux polariseurs [35]. Ces techniques ont une profondeur de champ limitée et la résolution radiale varie le long de l’axe de propagation. Une alternative intéressante est d’utiliser une lentille conique appelée axicon (Figure 4) qui focalise la lumière sur une ligne d’excitation dans l’axe des z, éliminant ainsi la nécessité d’effectuer des balayages successifs afin d’imager tout le volume d’intérêt. Cet élément optique, compatible avec la M2P, focalise la lumière sur une mince ligne axiale tout en maintenant une résolution radiale comparable à celle d’un objectif conventionnel (Figure 4). On a montré qu’en M2P, un microscope à base d’axicon permet d’obtenir, en un seul balayage, des images fonctionnelles de structures distribuées à différentes profondeurs focales comparables à la somme d’images obtenues avec une lentille conventionnelle [36, 37].
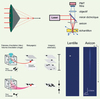 |
Figure 4. Un microscope à deux photons utilisant un axicon pour maximiser la profondeur de champ sans compromettre la résolution radiale. A. Schématisation d’un axicon d’angle α. La lumière est focalisée sur une ligne d’excitation qui traversera le plan xy du spécimen à imager.B. Schéma expérimental du montage de la M2P utilisant un axicon. Dans ce cas, c’est l’axicon qui est déplacé transversalement afin de balayer l’échantillon. Le même axicon est utilisé pour récolter la fluorescence émise et la rediriger vers le système de détection. C. Illustration du volume d’excitation et des images résultant du balayage d’un échantillon avec un objectif (haut) et avec un axicon (bas). D. Simulations montrant le profil d’intensité au carré pour une lentille (gauche) et un axicon (droite) (ouverture numérique = 0,16 et coefficient d’absorption = 8,5 cm-1). Pour un axicon de 30°, la ligne focale obtenue est d’une longueur de l’ordre du mm tout en ayant une résolution axiale constante de l’ordre du micron. |
Autres applications
Au-delà de l’imagerie, les nombreux avantages de la M2P par rapport à la microscopie confocale (Tableau I) ouvrent la porte à des applications intéressantes pour le traitement de certaines pathologies, notamment sous forme de thérapie dite « photodynamique ». On vise ici à détruire des tissus spécifiques, comme par exemple des tumeurs ou des excroissances. Ces tissus sont au préalable chargés d’un agent photosensible qui, une fois photo activé, devient toxique pour les cellules. Les mêmes avantages de l’optique non-linéaire que ceux présentés ci-dessus pour l’imagerie peuvent être exploités pour cibler le photo-dommage de manière très spécifique : à une profondeur donnée et dans un volume très précis [38].
Comparaison entre la microscopie confocale et la microscopie à deux photons.

Article reçu le 7 juin 2006, accepté le 28 juin 2006.
Références
- Göppert-Mayer M. Uber elementarakte mit zwei quanten-sprüngen. Ann Phys 1931; 9 : 273–95. [Google Scholar]
- Kaiser W, Garrett CGB. Two-photon excitation in CaF2Eu2+. Phys Rev Lett 1961; 7 : 229–31. [Google Scholar]
- Peticolas WL, Goldsborough JP, Rieckhoff KE. Double photon excitation in organic crystals. Phys Rev Lett 1963; 10 : 43–5. [Google Scholar]
- Sheppard CJR, Kompfner R. Resonant scanning optical microscope. Appl Opt 1978; 17 : 2879–82. [Google Scholar]
- Denk W, Strickler JH, Webb WW. Two-photon laser scanning fluorescence microscopy. Science 1990; 248 : 73–6. [Google Scholar]
- Davidovits P, Egger MD. Scanning laser microscope. Nature 1969; 233 : 831. [Google Scholar]
- Xu C, Guild J, Webb W, Denk W. Determination of absolute two-photon excitation cross sections by in situ second-order autocorrelation. Opt Lett 1995; 20 : 2372. [Google Scholar]
- Xu C, Webb WW. Measurement of two-photon excitation cross sections of molecular fluorophores with data from 690 to 1050 nm. J Opt Soc Am B Optic Physics 1996; 13 : 481. [Google Scholar]
- Schilders SP, Gu M. Limiting factors on image quality in imaging through turbid media under single-photon and two-photon excitation. Microsc Microanal 2000; 6 : 156–60. [Google Scholar]
- Westphal V, Hell SW. Nanoscale resolution in the focal plane of an optical microscope. Phys Rev Lett 2005; 94 : 143903/1–4. [Google Scholar]
- Willig KI, Rizzoli SO, Westphal V, et al. STED microscopy reveals that synaptotagmin remains clustered after synaptic vesicle exocytosis. Nature 2006; 440 : 935–9. [Google Scholar]
- Hell SW, Lindek S, Cremer C, Stelzer EHK. Measurement of the 4Pi-confocal point spread function proves 75 nm axial resolution. Appl Phys Lett 1994; 64 : 1335–7. [Google Scholar]
- Hell SW, Dyba M, Jakobs S. Concepts for nanoscale resolution in fluorescence microscopy. Curr Opin Neurol 2004; 14 : 599–609. [Google Scholar]
- Varin C, Piché M. Analytical calculation of the longitudinal electric field resulting from the tight focusing of an ultrafast transverse magnetic beam. J Opt Soc Am A 2006 (sous presse). [Google Scholar]
- Zipfel W, Williams RM, Webb WW. Nonlinear magic: multiphoton microscopy in the biosciences. Nat Biotech 2003; 21 : 1369–77. [Google Scholar]
- Helmchen F, Denk W. Deep tissue two-photon microscopy. Nat Methods 2005; 2 : 932–40. [Google Scholar]
- Wang H, Backus S, Chang Z, et al. Generation of 10-W average-power, 40-TW peak-power, 24-fs pulses from a Ti:sapphire amplifier system. J Opt Soc Am B 1999; 16 : 1790–4. [Google Scholar]
- Beaurepaire E, Oheim M, Mertz J. Ultra-deep two-photon fluorescence excitation in turbid media. Opt Commun 2001; 188 : 25–9. [Google Scholar]
- Theer P, Hasan MT, Denk W. Two-photon imaging to a depth of 1000 microns in living brains by use of a Ti:Al2O3 regenerative amplifier. Opt Lett 2003; 28 : 1022–4. [Google Scholar]
- Cognet L, Groc L, Lounis V, Choquet D. Multiple routes for glutamate receptor trafficking: surface diffusion and membrane traffic cooperate to bring receptors to synapses. Sci STKE 2006; 2006 : pe13. [Google Scholar]
- Larson DR, Zipfel WR, Williams RM, et al. Water-soluble quantum dots for multiphoton fluorescence imaging in vivo. Science 2003; 300 : 1434–6. [Google Scholar]
- Wiseman PW, Squier JA, Ellisman MH, Wilson KR. Two-photon image correlation spectroscopy and image cross-correlation spectroscopy. J Microsc 2000; 200 : 14–25. [Google Scholar]
- Mehta AD, Jung JC, Flusberg BA, Schnitzer. Fiber optic in vivo imaging in the mammalian nervous system. Curr Opin Neurobiol 2004; 14 : 1–12. [Google Scholar]
- Sakmann B, Neher E. Single-channel recording. New York : Plenum Press, 1995. [Google Scholar]
- Yuste R, Denk W. Dentritic spines as basic functional units of neuronal integration. Nature 1995; 375 : 682–4. [Google Scholar]
- Oertner T, Sabatini BL, Nimchinsky EA, Svoboda K. Facilitation at single synapses probed with optical quantal analysis. Nat Neurosci 2002; 5 : 657–64. [Google Scholar]
- Yasuda R, Nimchinsky EA, Scheuss V, et al. Imaging calcium concentration dynamics in small neuronal compartments. Sci STKE 2004; 2004 : l5. [Google Scholar]
- Tsien R, Bacskai B. Video-rate confocal microscopy. In : Palwley JB, ed. Handbook of biological confocal microscopy. New York : Plenum Press, 1995 : 459–78. [Google Scholar]
- Salomé R, Kremer Y, Dieudonne S, et al. Ultrafast random-access scanning in two-photon microscopy using acousto-optic deflectors. J Neurosci Methods 2006 : 154 :161–74. [Google Scholar]
- Stosiek C, Garaschuk O, Holthoff K, Konnerth A. In vivo two-photon calcium imaging of neuronal networks. Proc Natl Acad Sci USA 2003; 100 : 7319–24. [Google Scholar]
- Cossart R, Aronov D, Yuste R. Attractor dynamics of network UP states in the neocortex. Nature 2003; 423 : 283–8. [Google Scholar]
- Cossart R, Ikegaya Y, Yuste R. Calcium imaging of cortical networks dynamics. Cell Calcium 2005; 37 : 451–7. [Google Scholar]
- Fittinghoff DN, Wisemann PW, Squier JA. Widefield multiphoton and temporally decorrelated multifocal multiphoton microscopy. OSA 2000; 7 : 273–9. [Google Scholar]
- Davidson N, Friesem AA, Hasman E. Holographic axilens : high resolution and long focal depth. Opt Lett 1991; 16 : 523–5. [Google Scholar]
- Sanyal S, Ghosh A. High focal depth with a quasi-bifocus birefringent lens. Appl Opt 2000; 39 : 2321–5. [Google Scholar]
- Dufour P, McCarthy N, De Koninck Y. Imaging of biological tissues: overview of optical configurations for linear and two-photon microscopy. Physics in Canada 2006; 62 : 75–82. [Google Scholar]
- Dufour P, Piché M, De Koninck Y, McCarthy N. Two-photon excitation fluorescence microscopy with high depth of field using an axicon. Appl Opt 2006 (sous presse). [Google Scholar]
- Wilson BC. Photodynamic therapy. In : Advances in biophotonics. NATO science series. Amsterdam : IOS Press, 2005. [Google Scholar]
Liste des tableaux
Comparaison entre la microscopie confocale et la microscopie à deux photons.
Liste des figures
|
Figure 1. Principes de microscopie multiphotonique. A. Schématisation des niveaux d’énergie d’une molécule fluorescente. La flèche bleue représente l’absorption à un photon (1p), les flèches rouges représentent l’absorption à 2 photons (2p) et les grises représentent l’absorption à 3 photons (3p). La flèche pointillée représente le passage vers un niveau d’énergie intermédiaire avant l’émission spontanée d’un photon de fluorescence (flèche verte). B. Schéma expérimental du montage de la M2P conventionnelle. La source d’excitation est dirigée vers l’échantillon via un système de balayage et un miroir dichroïque, puis elle est concentrée au point focal d’excitation par l’objectif. La fluorescence émise par l’échantillon est ensuite collectée par le même objectif, filtrée par un filtre d’émission et détectée par un tube photomultiplicateur (PMT). Le volume d’excitation doit être éclairé pendant un certain temps (appelé pixel dwell time) afin que l’on puisse récolter assez de photons de fluorescence pour produire un signal clair. |
| Dans le texte | |
|
Figure 2. Avantages et limites de la M2P pour la résolution spatiale. A. Images 2D du profil d’intensité lumineuse de points focaux (1P ; dans le plan xz ou yz à 450 et 800 nm, respectivement) et intensité au carré à 800 nm (2P) obtenus par simulation. L’ouverture numérique de l’objectif est de 0,5 et le coefficient d’absorption est nul. La perte de résolution en utilisant une longueur d’onde plus grande (la résolution est proportionnelle à la largeur à mi-hauteur des points focaux) se trouve compensée par le fait que l’absorption de 2P est proportionnelle à l’intensité au carré. Le résultat net est que la résolution radiale avec une excitation à 2P à 800 nm est comparable à celle obtenue avec 1P à 450 nm. B. Coefficient d’absorption des principaux absorbeurs dans les tissus en fonction de la longueur d’onde. Notez l’absorption 10 à 100 fois plus faible à 800 nm qu’à 450 nm (flèches) due à la présence d’éléments absorbant dans le tissu (mélamine, hémoglobine…). C. Profil de l’intensité lumineuse à 1P (gauche) et profil de l’intensité au carré à 2P (droite) obtenus par simulation en tenant compte, cette fois-ci, du coefficient d’absorption. La valeur du coefficient d’absorption est de 85 cm-1 dans le cas à 1P et de 8,5 cm-1 dans le cas à 2P (ouverture numérique = 0,16). D. Image d’un neurone pyramidal du néocortex exprimant la green fluorescent protein (GFP) à faible grossissement à gauche et avec un grossissement croissant vers la droite pour illustrer le détail des prolongements du neurone : le panneau du milieu met en évidence une dendrite ; le panneau de droite les détails d’épines dendritiques. Comme les épines ont une taille de l’ordre du micromètre, la résolution optique standard ne permet pas de distinguer leur structure fine. E. Exemple de technique utilisée pour augmenter la résolution en deçà des limites de diffraction de la lumière : la STED (stimulated emission depletion). On excite l’échantillon avec une impulsion laser de la même façon qu’on le fait en M2P. Par la suite, avant que les fluorophores réémettent la fluorescence, on désexcite les molécules autour du point focal par émission stimulée avec une impulsion en forme de beigne. Le volume d’excitation correspond alors à une soustraction entre le volume d’excitation et le volume de désexcitation autour du point focal. Les molécules encore excitées sont alors dans un volume beaucoup plus petit que le point d’excitation engendré par l’excitation, ce qui améliore la résolution radiale. (E1) présente le profil d’intensité de l’impulsion laser d’excitation, (E2) le profil d’intensité du faisceau en beigne qui suit l’impulsion d’excitation. (E3) montre le profil d’excitation résultant après le passage des deux premières impulsions, (E4) le profil inverse du beigne. |
| Dans le texte | |
|
Figure 3. Imagerie fonctionnelle en profondeur dans le tissu. A. Image reconstituée de cellules corticales dans le cerveau intact de souris transgéniques exprimant une protéine fluorescente dans une sous-population de neurones corticaux de la couche V. Une projection sur le côté de l’empilement des images permet de visualiser les cellules dans l’axe des z et d’illustrer la capacité d’imager en profondeur dans la matière grise du cortex cérébral (tirée de [16]). B. Graphique de l’intensité en fonction du temps pour un faisceau laser continu (noir) et pour des faisceaux laser pulsés à 100 MHz (bleu), à 10 MHz (vert) et à 250 kHz (rouge). Les faisceaux à 10 MHz et 250 kHz sont engendrés à l’aide d’un amplificateur régénératif. Notons que les quatre faisceaux laser ont la même puissance moyenne (correspondant à la puissance du faisceau continu). C. Imagerie fonctionnelle sur un bouton synaptique. (C1) Schéma de la circuiterie de l’hippocampe chez le rat. Les cellules granulaires (CG) sont situées dans le gyrus denté et envoient leur prolongement axonal dans la région CA3 où ils font synapse au niveau de dendrites proximales des cellules pyramidales. Les boutons synaptiques des CG peuvent être marqués à l’aide d’un indicateur calcique qui servira à rapporter les changements calciques de cette terminaison suite à une stimulation électrique des axones. (C2) Terminaisons en boutons marquées à l’aide de l’indicateur calcique Oregon Green BAPTA1-AM. La terminaison imagée est encadrée en pointillés. (C3) Pour maximiser la résolution temporelle on ne balaye qu’une seule ligne qui passe à travers le bouton. (C4) Les changements de fluorescence sont rapportés sous forme graphique dans le temps. D. Imagerie fonctionnelle d’un large réseau de neurones. (D1) Image en M2P de fluorescence provenant d’un marqueur calcique capturé par un grand nombre de neurones corticaux (plusieurs centaines de neurones peuvent être suivis avec cette méthode). (D2) Exemple de tracés rapportant la fluctuation de fluorescence dans le temps dans deux des neurones du champ en d1. (D3) Ces fluctuations peuvent être comptées à partir d’un seuil de détection donné et (D4) l’activité du réseau peut être reconstituée tel que sur ce raster-plot où chaque ligne représente un neurone (modifié à partir de [31]). |
| Dans le texte | |
|
Figure 4. Un microscope à deux photons utilisant un axicon pour maximiser la profondeur de champ sans compromettre la résolution radiale. A. Schématisation d’un axicon d’angle α. La lumière est focalisée sur une ligne d’excitation qui traversera le plan xy du spécimen à imager.B. Schéma expérimental du montage de la M2P utilisant un axicon. Dans ce cas, c’est l’axicon qui est déplacé transversalement afin de balayer l’échantillon. Le même axicon est utilisé pour récolter la fluorescence émise et la rediriger vers le système de détection. C. Illustration du volume d’excitation et des images résultant du balayage d’un échantillon avec un objectif (haut) et avec un axicon (bas). D. Simulations montrant le profil d’intensité au carré pour une lentille (gauche) et un axicon (droite) (ouverture numérique = 0,16 et coefficient d’absorption = 8,5 cm-1). Pour un axicon de 30°, la ligne focale obtenue est d’une longueur de l’ordre du mm tout en ayant une résolution axiale constante de l’ordre du micron. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.