")
")
| Issue |
Med Sci (Paris)
Volume 22, Number 4, Avril 2006
|
|
|---|---|---|
| Page(s) | 411 - 415 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/2006224411 | |
| Published online | 15 avril 2006 | |
Les sphingolipides : vecteurs d’agents pathogènes et cause de maladies génétiques
Sphingolipids, vehicle for pathogenic agents and cause of genetic diseases
1
Département de Pharmacologie, Faculté de Médecine, Université de Montréal, H3C 3J7 Montréal, Québec, Canada
2
Laboratoire de Chimie Biologique Appliquée, UMR-INRA 1111, Université Paul Cézanne, 13397 Marseille, France
3
Laboratoire de Physiologie Neurovégétative, UMR CNRS 6153-INRA 1147, Université Paul Cézanne, 13397 Marseille, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Les sphingolipides (SPL) sont des molécules ubiquitaires indispensables au maintien et au développement des organismes vivants. Ils ne sont pas répartis uniformément le long de la membrane mais regroupés sous forme de microdomaines lipidiques appelés rafts. On a longtemps pensé que les SPL avaient uniquement un rôle structural. On sait maintenant qu’ils jouent aussi un rôle de récepteur et de seconds messagers (intervenant dans des fonctions majeures de la vie cellulaire) et que de nombreuses maladies génétiques (sphingolipidoses) s’expliquent par un dysfonctionnement de leur métabolisme. Après un rappel des propriétés structurales des sphingolipides, cet article fait le point sur les relations entre leur rôle de récepteur et l’entrée d’agents pathogens dans la cellule ainsi que sur les mécanismes pathologiques des sphingolipidoses.
Abstract
Sphingolipids are present in all eukaryotic cells and share a sphingoid base : sphingosine. They were first discovered in 1884 and for a long time they were thought to participate to membrane structure only. Recently it has been established that they are mainly located in particular areas of the membrane called rafts which are signalling platforms. It has also been demonstrated that sphingolipids are receptors and second messengers. They play a crucial role in cellular functioning and are necessary to maintenance and developing of living organisms. However due to their receptor properties, they are also gateway for penetration of pathogenic agents such as virus (Ebola, HIV) or toxins (botulinium, tetanus). These agents first bind to glycosphingolipids or proteins mainly located in rafts. The complex so formed is required for the crossing of the membrane by the pathogenic agent. Sphingolipids metabolism is regulated by numerous enzymes. A failure in the activity of one of them induces an accumulation of sphingolipids known as sphingolipidoses. These are genetic diseases having severe consequences for the survival of the organism. The precise mechanisms of the sphingolipidoses are still mainly unknown which explains why few therapeutic strategies are available. These particular properties of lipids rafts and sphingolipids explain why a growing number of studies in the medical and scientific fields are devoted to them.
© 2006 médecine/sciences - Inserm / SRMS
En 1884, un composé lipidique a été isolé à partir de tissus cérébraux. Sa structure et son métabolisme étant inconnus, il fut nommé sphingomyéline en référence au sphinx, animal énigmatique. Actuellement, plus de 300 sphingolipides (SPL) ont été identifiés : ce sont des constituants de la membrane plasmique. Les SPL sont des molécules ubiquitaires présentes chez les cellules eucaryotes et certaines bactéries [1] ; ils ont une base sphingoïde commune, la sphingosine (Figure 1). Lorsque la sphingosine est liée à un acide gras par une liaison amide, on obtient une sphingosine N-acylée, communément appelée céramide. L’acide gras constitutif du céramide est une chaîne carbonée, généralement saturée, contenant de 14 à 26 atomes de carbone. Ces deux chaînes carbonées (sphingosine et acide gras) forment la queue apolaire des SPL, leur tête polaire étant représentée par des sucres ou un groupement phosphate lié à un alcool. En fonction de la nature de leur tête polaire, le groupe des SPL se divise en deux sous-groupes : les glycosphingolipides et les sphingophospholipides [2]. Les glycosphingolipides ont pour tête polaire un groupement glucidique. Ils sont représentés par les cérébrosides, les sulfatides et les gangliosides. Chez les mammifères, les sphingophospholipides sont représentés par la sphingomyéline et ses dérivés : céramide-1-phosphate, sphingosine-1-phosphate et sphingosylphosphorylcholine [3] (Figure 2). La tête polaire des sphingomyélines est un groupement phosphate lié à un alcool, la choline [4]. Les SPL peuvent jouer un rôle structural, posséder une activité de récepteurs et se comporter comme des seconds messagers (Figure 2). Dans cet article, seuls les aspects liés aux propriétés structurales et de récepteurs seront abordés.
 |
Figure 1. La sphingosine, base commune de tous les sphingolipides. Ce lipide est constitué d’une chaîne à 18 atomes de carbones. Celle-ci porte deux groupements OH en position C1 et C3, un groupement NH2 en position C2 et une insaturation (double liaison) entre C4 et C5. L’ajout d’un acide gras par liaison amide forme alors un céramide. Si une tête polaire se lie au céramide par le groupement OH en position C1, on obtient un glycosphingolipide ou un sphingophospholipide. |
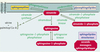 |
Figure 2. Voies de production des seconds messagers sphingolipidiques. Elles sont au nombre de trois : une voie de novo et deux voies d’hydrolyse. La voie de novo comporte l’association d’une molécule de sérine et de palmitoyl Co-A dans le réticulum endoplasmique, ce qui aboutit à la formation de céramide. Les voies d’hydrolyse produisent du céramide respectivement à partir de la sphingomyéline et des glycosphingolipides. Toutes ces transformations sont réversibles, elles sont catalysées par des enzymes spécifiques : sphingomyélinases, cérébrosidases, céramidases, kinases et phosphatases. |
Sphingolipides et organisation de la membrane cellulaire
Les composants membranaires sont distribués en microdomaines hétérogènes ou rafts qui se caractérisent par leur richesse en cholestérol, SPL et protéines spécifiques [5]. Ces rafts interviennent dans des processus cellulaires majeurs : signalisation, régulation du cholestérol ou trafic membranaire [6–13].
Chez les mammifères, les SPL représentent une partie importante (environ 20 %) des lipides du système nerveux. La sphingomyéline y est particulièrement abondante et peut représenter plus de 6 % des lipides totaux d’un neurone [14]. Les glycosphingolipides sont indispensables au processus de myélinisation des fibres nerveuses. Une perturbation dans la biosynthèse de ces lipides provoque, pour les neurones myélinisés, la perte de la conduction saltatoire du potentiel d’action [15].
La sphingomyéline joue également un rôle cytoprotecteur important en maintenant l’homéostasie de la membrane plasmique [16]. Les propriétés structurales de la sphingomyéline lui confèrent, en effet, trois fonctions protectrices complémentaires contre les agressions externes : protection des glycérophospholipides membranaires d’une hydrolyse prématurée et excessive, diminution de la fluidité membranaire qui entrave la propagation des radicaux peroxydes à travers la bicouche lipidique et inhibe la peroxydation des lipides, enfin, protection des autres lipides membranaires contre les agents oxydants en agissant comme une molécule isolante.
Rôle de récepteur des sphingolipides
La fonction de récepteur des SPL est assurée par les glycosphingolipides qui sont exclusivement situés dans le feuillet extracellulaire [17]. Les sucres que portent les glycosphingolipides sont des motifs de reconnaissance impliqués dans des fonctions physiologiques, infectieuses ou pathologiques.
Le rôle des glycosphingolipides en tant que récepteur est aujourd’hui bien décrit lors de l’inhibition de la croissance axonale. La régénérescence axonale, survenant après une atteinte des nerfs périphériques, est un processus moteur où l’extrémité axonale en cours d’élongation exprime des récepteurs sensibles aux signaux environnementaux. En revanche, ce processus de régénérescence axonale n’existe pas dans le système nerveux central où l’inhibition de la croissance axonale est due à la présence de cellules gliales particulières, les oligodendrocytes. Trois protéines inhibitrices de la croissance axonale ont été isolées à partir de la myéline. Il s’agit des protéines MAG (myelin-associated glycoprotein), Nogo et OMgp (oligodendrocyte-myelin glycoprotein) [18]. Les premiers récepteurs de la protéine MAG qui ont été identifiés sont des SPL, les gangliosides GD1a et GT1b. Ils activent Rho, une petite protéine G ancrée dans le feuillet interne de la membrane, qui active à son tour la protéine Rho kinase responsable de l’inhibition de la croissance axonale [19].
Pour coloniser leurs cellules hôtes, de nombreux agents pathogènes (parasites, virus, bactéries) utilisent comme récepteur ou corécepteur les protéines et les lipides constitutifs des rafts (glycosphingolipides, sphingomyéline et cholestérol) [20]. Les toxines botuliques et tétaniques agissent sur les neurones en se liant aux gangliosides GD1a, GD1d et GT1b qui sont utilisés normalement dans les processus d’adhésion cellulaire. Cependant, ces toxines présentent peu d’affinité avec les glycosphingolipides alors que leur toxicité est très élevée, même pour de faibles concentrations. Pour expliquer ce paradoxe, Montecucco proposa la théorie du « double récepteur » qui fait intervenir deux étapes dans le processus d’internalisation des toxines. Les glycosphingolipides servent de premier récepteur en permettant la fixation des toxines par une liaison de faible affinité. Le complexe toxine/gangliosides se déplace ensuite latéralement pour se lier avec une protéine réceptrice située plus loin dans la membrane. Cette protéine joue le rôle de second récepteur. La forte toxicité peut s’expliquer de deux façons : soit la liaison de la toxine avec le ganglioside provoque un réarrangement conformationnel de la structure de la toxine qui augmente son affinité pour la protéine réceptrice, soit c’est la somme des deux liaisons faibles, toxine/gangliosides et toxine/protéine réceptrice, qui procure une forte affinité de la toxine pour sa cellule hôte [21, 22]. La toxine du choléra se fixe également sur les glycosphingolipides. Cette toxine pentavalente se lie avec cinq gangliosides de type GM1 par des liaisons de forte affinité. L’interaction toxine/gangliosides GM1 assure donc une liaison maximale de la toxine du choléra à la surface de la membrane afin qu’elle puisse entrer dans la cellule grâce à son peptide actif qui pourra alors activer l’adénylate cyclase [22]. De nombreux virus utilisent les rafts lipidiques comme voies d’infection de leurs cellules hôtes. C’est le cas du virus Ebola et du virus de Marburg [23], du virus influenza [24] et du virus de l’immunodéficience humaine (VIH) [22]. Parmi ces virus, le VIH utilise les glycosphingolipides comme corécepteur lors de l’infection. Pour entrer dans le milieu interne d’un organisme, le VIH se fixe sur les muqueuses où il reconnaît spécifiquement le ganglioside galactosylcéramide à la surface des cellules épithéliales. Puis le VIH est transporté par transcytose à travers l’épithélium pour être enfin libéré du côté basolatéral [24]. Une fois dans le milieu interne, il va infecter les cellules T CD4+ du système immunitaire. Le VIH se fixe à leur membrane en reconnaissant des protéines (CD4) et des SPL (ganglioside GM3, globoside Gb3) constitutifs des rafts des cellules T CD4+. Il crée alors un complexe trimoléculaire où la fonction des glycosphingolipides est de stabiliser le VIH à la surface cellulaire puis de faciliter la migration de ce complexe vers un corécepteur. Le virus fusionne alors avec la membrane plasmique des cellules T pour y introduire son ARN génomique et se reproduire.
Les SPL sont également impliqués dans des maladies auto-immunes. En effet, plus de 20 glycosphingolipides sont les cibles d’auto-anticorps responsables, chez l’homme, de nombreuses neuropathies dont le syndrome de Guillain-Barré [25]. C’est une atteinte nerveuse périphérique avec ataxie et perte des réflexes tendineux. Ce syndrome se caractérise par la présence d’auto-anticorps anti-GM1 ou -GM2 [26].
Sphingolipides et maladies génétiques
Il existe de nombreuses maladies dues à un dysfonctionnement du métabolisme des SPL. Celles-ci sont principalement d’origine génétique et concernent leur biosynthèse ou leur dégradation (sphingolipidoses). Les sphingolipidoses se transmettent selon le mode autosomique récessif [27]. Elles ont pour cause une déficience dans l’activité d’enzymes situées dans les lysosomes et intervenant dans la dégradation des SPL. Les sphingolipidoses ou maladies lysosomiales se caractérisent très souvent par l’accumulation d’un métabolite sphingolipidique qui est détectable dans les cellules par des techniques de coloration ou des études ultrastructurales. La sévérité de ces maladies est d’autant plus grande qu’elles apparaissent précocement. Nous présentons seulement ici quelques affections caractéristiques des principaux types de sphingolipidoses (Figure 3). Nous abordons ensuite le problème des mécanismes physiopathologiques mis en jeu et de leurs conséquences.
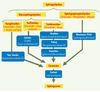 |
Figure 3. Principales sphingolipidoses. Celles-ci se caractérisent par l’accumulation d’un sphingolipide (SPL). Les sphingolipidoses sont classées en plusieurs catégories selon la nature du SPL accumulé : gangliosides (maladie de Tay-Sachs), sulfatides (leucodystrophie métachromatique : LDM ou maladie de Scholz), cérébrosides (maladies de Krabbe, Fabry et Gaucher), sphingophospholipides (maladie de Niemann-Pick) et enfin, céramide (maladie de Farber). L’enzyme déficiente responsable de cette accumulation est indiquée sous le nom de la maladie. |
Les sphingomyélinoses sont caractérisées par une accumulation de sphingophospholipides. La maladie de Niemann-Pick est due à un déficit en sphingomyélinase acide qui provoque une accumulation de sphingomyéline [28]. On en distingue plusieurs types : les types A et B sont dus à des mutations du gène de la sphingomyélinase qui est porté par le chromosome 11, alors que le type C est dû à un dysfonctionnement du métabolisme du cholestérol. Le type A se caractérise par une atteinte neurodégénérative et une organomégalie affectant le foie et la rate. Le type B est non neurodégénératif et se caractérise principalement par une hépatosplénomégalie.
Les sphingoglycolipidoses se caractérisent par une accumulation de glycosphingolipides qui peuvent être des cérébrosides, des sulfatides ou des gangliosides. La maladie de Gaucher est due à un déficit en glucocérébrosidase qui est l’enzyme de dégradation du glucocérébroside en glucose et céramide [29]. Il en résulte une accumulation de glucocérébrosides dans la rate, la moelle osseuse et le système nerveux. Le type 1 ou adulte est non neuropathique ; il est caractérisé par une hépatosplénomégalie, une thrombopénie, une anémie, un retard de croissance et une pathologie osseuse de type ostéonécrose. Le type 2 ou infantile se caractérise par une atteinte du système nerveux à évolution rapide se manifestant par un arrêt du développement psychomoteur, une hypertonie et des convulsions. Dans la maladie de Fabry, un déficit en α-galactosidase A induit une accumulation de trihexosylcéramide dans les cellules endothéliales, cardiaques et rénales [30]. La maladie de Krabbe est due à un déficit en galactocérébrosidase, le gène déficient étant localisé sur le chromosome 14. Cette déficience enzymatique produit dans les oligodendrocytes une accumulation anormale d’un dérivé du galactocérébroside, la galactosylsphingosine [31]. Les oligodendrocytes, qui sont à l’origine de la formation des gaines de myéline du système nerveux central, sont alors détruits.
Les sphingolipidoses sulfatiques sont caractérisées par une accumulation de sulfatides. La maladie de Scholz-Greenfield (ou leucodystrophie métachromatique) est due à un déficit en aryl-sulfatase A dont le gène est situé sur le chromosome 22. Ce déficit provoque une accumulation de sulfatides dans le système nerveux central et périphérique et affecte la conduction nerveuse [32].
Les gangliosidoses sont caractérisées par une accumulation de gangliosides. La maladie de Tay-Sachs est due à un déficit en hexosaminidase A qui entraîne une accumulation de gangliosides GM2 [33].
La maladie de Farber, distincte des types précédents, est caractérisée par un déficit de la céramidase acide, responsable d’une accumulation de céramide [34] qui provoque des nodules sous-cutanés, des atteintes neurologiques et une hépatosplénomégalie.
Selon l’hypothèse la plus couramment admise, c’est la surcharge en SPL elle-même qui perturbe le fonctionnement cellulaire. Cela peut se concevoir dans le cas de molécules biologiquement inactives. Cependant, le mécanisme physiopathologique pourrait être lié à l’activité de certaines molécules [35]. Dans la maladie de Krabbe, le galactocérébroside est dégradé par une voie catabolique accessoire en une substance, normalement produite à l’état de trace, la galactosylsphingosine (psychosine). Cette substance se comporte comme un médiateur lipidique qui active des récepteurs orphelins TDAG8 couplés à des protéines G, ce qui provoque l’apoptose des oligodendrocytes [36, 37]. Par ailleurs, il est maintenant connu que certains SPL sont des seconds messagers qui ont un rôle majeur dans le fonctionnement cellulaire. L’équilibre entre les concentrations de certains d’entre eux (céramide et sphingosine-1-phosphate, par exemple) joue le rôle d’un véritable rhéostat qui conditionne le devenir cellulaire : prolifération ou apoptose [38]. Dans ces conditions, un déséquilibre majeur dans les concentrations de ces molécules pourra avoir des conséquences drastiques pour la cellule. Une étude effectuée chez l’homme indique effectivement que les phénomènes pro-apoptotiques décrits dans la maladie de Farber seraient dus au céramide [39]. En fait, l’étude des mécanismes des sphingolipidoses en est à ses débuts puisqu’on commence seulement à disposer de modèles animaux présentant une accumulation de sphingolipides. Il a ainsi été démontré sur un modèle de rat obèse diabétique (Zucker diabetic fatty rats) que les cellules cardiaques subissent une apoptose plus importante que chez des rats témoins de type sauvage. Ce processus est dû, entre autres, à une production anormale de céramide à l’état basal [40]. Le faible niveau de connaissances dont nous disposons actuellement concernant les mécanismes des sphingolipidoses explique qu’il existe encore peu de traitements efficaces. La plupart sont en effet de type palliatif. Les résultats les plus encourageants sont obtenus par substitution d’enzyme recombinante, glucosidase ou sphingomylinase [41–43], et dans une moindre mesure, par diminution du substrat [44–47]. L’avenir réside dans la mise au point de thérapies géniques visant à réintroduire le gène déficient [48].
Conclusions et enjeux actuels
Les SPL sont principalement contenus dans des zones particulières de la membrane appelées rafts qui font l’objet de travaux de recherche intensifs. Les connaissances ainsi acquises ont permis des avancées décisives dans la compréhension des mécanismes mis en jeu par certains agents pathogènes lors de leur pénétration dans la cellule. On sait maintenant que les rafts sont la cible privilégiée de ces agents. En effet, certaines molécules constitutives des rafts (et en particulier les SPL) servent de ligands indispensables à la fixation de ces agents pathogènes. Par ailleurs, un dysfonctionnement dans le métabolisme des SPL est à l’origine de maladies sévères d’origine génétique, les sphingolipidoses. L’analyse des mécanismes responsables de ces maladies en est encore à ses débuts ce qui explique que l’on dispose de peu de traitements efficaces. Le rôle des SPL dans l’homéostasie cellulaire explique qu’ils soient l’objet d’un nombre sans cesse croissant d’études effectuées dans des disciplines médicales et scientifiques. L’enjeu de ces études est, à terme, une meilleure compréhension du rôle de la membrane plasmique et une efficacité accrue dans la lutte contre certaines maladies infectieuses ou génétiques.
Remerciements
Les travaux provenant de nos laboratoires ont été financés par l’Université Paul Cézanne (Aix-marseille III), le CNRS et L’INRA.
Article reçu le 19 septembre 2005, accepté le 5 décembre 2005.
Références
- Karlsson KA. Sphingolipid long chain bases. Lipids 1970; 5 : 878–91. [Google Scholar]
- Liu G, Kleine L, Hebert R. Advances in the signal transduction of ceramide and related sphingolipids. Crit Clin Lab Sci 1999; 36 : 511–73. [Google Scholar]
- Rietveld A, Simons K. The differential miscibility of lipids as the basis for the formation of functional membrane rafts. Biochim Biophys Acta 1998; 1376 : 467–79. [Google Scholar]
- Ramstedt B, Slotte JP. Membrane properties of sphingomyelins. FEBS Lett 2002; 531 : 33–7. [Google Scholar]
- Brown DA, London E. Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J Biol Chem 2000; 275 : 17221–4. [Google Scholar]
- Simons K, van Meer G. Lipid sorting in epithelial cells. Biochemistry 1988; 27 : 6197–202. [Google Scholar]
- Simons K, Ikonen E. Functional rafts in cell membranes. Nature 1997; 387 : 569–72. [Google Scholar]
- Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol 2000; 1 : 31–9. [Google Scholar]
- Hoessli DC, Ilangumaran S, Soltermann A, et al. Signaling through sphingolipid microdomains of the plasma membrane: the concept of signaling plateform. Glycoconj J 2000; 17 : 191–7. [Google Scholar]
- Tsui-Pierchala BA, Encinas M, Milbrandt J, Johnson EM Jr. Lipid rafts in neuronal signaling and function. Trends Neurosci 2002; 25 : 412–7. [Google Scholar]
- Pike LJ. Lipid rafts : bringing order to chaos. J Lipid Res 2003; 44 : 655–67. [Google Scholar]
- Hiol A, Davey PC, Osterhout JL, et al. Palmitoylation regulates RGS16 function I. Mutation of amino terminal cysteine residues on RGS16 prevents its targeting to lipid rafts and palmitoylation of an internal cysteine residue. J Biol Chem 2003; 278 : 19301–8. [Google Scholar]
- Osterhout JL, Waheed AA, Hiol A, et al. Palmitoylation regulates RGS16 function II. Palmitoylation of a cysteine residue in the RGS box is critical for RGS16 GTPase accelerating activity and regulation of Gi-coupled signalling. J Biol Chem 2003; 278 : 19309–16. [Google Scholar]
- O’Brien JS, Sampson EL. Lipid composition of the normal human brain : gray matter, white matter, and myelin. J Lipid Res 1965; 6 : 537–44. [Google Scholar]
- Stoffel W, Bosio A. Myelin glycolipids and their functions. Curr Opin Neurobiol 1997; 7 : 654–61. [Google Scholar]
- Subbaiah PV, Sargis RM. Sphingomyelin : a natural modulator of membrane homeostasis and inflammation. Med Hypotheses 2001; 57 : 135–8. [Google Scholar]
- Fishman PH, Brady RO. Biosynthesis and function of gangliosides. Science 1976; 194 : 906–15. [Google Scholar]
- McKerracher L, Winton MJ. Nogo on the go. Neuron 2002; 36 : 345–8. [Google Scholar]
- Yang LJ, Zeller CB, Shaper NL, et al. Gangliosides are neuronal ligands for myelin-associated glycoprotein. Proc Natl Acad Sci USA 1996; 93 : 814–8. [Google Scholar]
- Brown DA, London E. Functions of lipid rafts in biological membranes. Annu Rev Cell Dev Biol 1998; 14 : 111–36. [Google Scholar]
- Montecucco C. How do tetanus and botulinum toxins bind to neuronal membranes ? Trends Biochem Sci 1986; 11 : 314–7. [Google Scholar]
- Fantini J, Garmy N, Mahfoud R, Yahi N. Lipid rafts : structure, function and role in HIV, Alzheimers and prion diseases. Expert Rev Mol Med 2002; 1–22. [Google Scholar]
- Bavari S, Bosio CM, Wiegand E, et al. Lipid raft microdomains : a gateway for compartmentalized trafficking of Ebola and Marburg viruses. J Exp Med 2002; 195 : 593–602. [Google Scholar]
- Simons K, Ehehalt R. Cholesterol, lipid rafts, and disease. J Clin Invest 2002; 110 : 597–603. [Google Scholar]
- O’Hanlon GM, Bullens RW, Plomp JJ, Willison HJ. Complex gangliosides as autoantibody targets at the neuromuscular junction in Miller Fisher syndrome : a current perspective. Neurochem Res 2002; 27 : 697–709. [Google Scholar]
- Hanada K. Sphingolipids in infectious diseases. Jpn J Infect Dis 2005; 58: 131–48. [Google Scholar]
- Futerman AH, van Meer G. The cell biology of lysosomal storage disorders. Nat Rev Mol Cell Biol 2004; 5 : 554–65. [Google Scholar]
- Kolodny EH. Niemann-Pick disease. Curr Opin Hematol 2000; 7 : 48–52. [Google Scholar]
- Jmoudiak M, Futerman AH. Gaucher disease: pathological mechanisms and modern management. Br J Haematol 2005; 129 : 178–88. [Google Scholar]
- Masson C, Cisse I, Simon V, et al. Fabry disease : a review. Joint Bone Spine 2004; 71 : 381–3. [Google Scholar]
- Suzuki K. Globoid cell leucodystrophy (Krabbe’s disease): update. J Child Neurol 2003; 18 : 595–603. [Google Scholar]
- Gieselmann V, Franken S, Klein D, et al. Metachromatic leukodystrophy : consequence of sulphatide accumulation. Acta Paediatr 2003; 92 (suppl) : 74–9. [Google Scholar]
- Kaback MM, Desnick RJ. Tay-Sachs disease : from clinical description to molecular defect. Adv Genet 2001; 44 : 1–9. [Google Scholar]
- Bar J, Linke T, Ferlinz K, et al. Molecular analysis of acid ceramidase deficiency in patients with Farber disease. Hum Mutat 2001; 17 : 199–209. [Google Scholar]
- Vellodi A. Lysosomal storage disorders. Br J Haematol 2005; 128 : 413–31. [Google Scholar]
- Suzuki K. Twenty five years of the "psychosine hypothesis": a personal perspective of its history and present status. Neurochem Res 1998; 23 : 251–9. [Google Scholar]
- Mitchison TJ. Psychosine, cytokinesis and orphan receptors : unexpected connections. J Cell Biol 2001; 153 : F1–4. [Google Scholar]
- Spiegel S, Milstien S. Sphingosine-1-phosphate : an enigmatic signalling lipid. Nat Rev Mol Cell Biol 2003; 4 : 397–407. [Google Scholar]
- Farina F, Cappello F, Todaro M, et al. Involvement of caspase-3 and GD3 ganglioside in ceramide induced apotosis in Farber disease. J Histochem. Cytochem 2000; 48 : 57–62. [Google Scholar]
- Zhou YT, Grayburn P, Karim A, et al. Lipotoxic heart disease in obese rats : implications for human obesity. Proc Natl Acad Sci USA 2000; 97 : 1784–9. [Google Scholar]
- Miranda SR, He X, Simonaro CM, et al. Infusion of recombinant human acid sphingomyelinase into Niemann-Pick disease mice leads to visceral, but not neurological, correction of the pathophysiology. FASEB J 2000; 14 : 1988–95. [Google Scholar]
- Bae JS, Jang KH, Schuchman TR, et al. Comparative effects of recombinant acid sphingomyelinase administration by different routes in Niemann-Pick disease mice. Exp Anim 2004; 53 : 417–21. [Google Scholar]
- Brady RO. Enzyme replacement therapy : conception, chaos and culmination. Philos Trans R Soc Lond B Biol Sci 2003; 358 : 915–9. [Google Scholar]
- Platt FM, Jeyakumar M, Andersson U, et al. Inhibition of substrate synthesis as a strategy for glycolipid lysosomal storage disease therapy. J Inherit Metab Dis 2001; 24 : 275–90. [Google Scholar]
- Zimran A, Elstein D. Gaucher disease and the clinical experience with substrate reduction therapy. Philos Trans R Soc Lond B Biol Sci 2003; 358 : 961–6. [Google Scholar]
- Moyses C. Substrate reduction therapy : clinical evaluation in type 1 Gaucher disease. Philos Trans R Soc Lond B Biol Sci 2003; 358 : 955–60. [Google Scholar]
- Cox TM. Substrate reduction therapy for lysosomal storage diseases. Acta Paediatr 2005; 94 (suppl) : 69–75. [Google Scholar]
- Ellinwood NM, Vite CH, Haskins ME. Gene therapy for lysosomal storage diseases: the lessons and promise of animal models. J Gene Med 2004; 6 : 481–506. [Google Scholar]
Liste des figures
|
Figure 1. La sphingosine, base commune de tous les sphingolipides. Ce lipide est constitué d’une chaîne à 18 atomes de carbones. Celle-ci porte deux groupements OH en position C1 et C3, un groupement NH2 en position C2 et une insaturation (double liaison) entre C4 et C5. L’ajout d’un acide gras par liaison amide forme alors un céramide. Si une tête polaire se lie au céramide par le groupement OH en position C1, on obtient un glycosphingolipide ou un sphingophospholipide. |
| Dans le texte | |
|
Figure 2. Voies de production des seconds messagers sphingolipidiques. Elles sont au nombre de trois : une voie de novo et deux voies d’hydrolyse. La voie de novo comporte l’association d’une molécule de sérine et de palmitoyl Co-A dans le réticulum endoplasmique, ce qui aboutit à la formation de céramide. Les voies d’hydrolyse produisent du céramide respectivement à partir de la sphingomyéline et des glycosphingolipides. Toutes ces transformations sont réversibles, elles sont catalysées par des enzymes spécifiques : sphingomyélinases, cérébrosidases, céramidases, kinases et phosphatases. |
| Dans le texte | |
|
Figure 3. Principales sphingolipidoses. Celles-ci se caractérisent par l’accumulation d’un sphingolipide (SPL). Les sphingolipidoses sont classées en plusieurs catégories selon la nature du SPL accumulé : gangliosides (maladie de Tay-Sachs), sulfatides (leucodystrophie métachromatique : LDM ou maladie de Scholz), cérébrosides (maladies de Krabbe, Fabry et Gaucher), sphingophospholipides (maladie de Niemann-Pick) et enfin, céramide (maladie de Farber). L’enzyme déficiente responsable de cette accumulation est indiquée sous le nom de la maladie. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.