")
")
| Issue |
Med Sci (Paris)
Volume 22, Number 3, Mars 2006
|
|
|---|---|---|
| Page(s) | 301 - 307 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/2006223301 | |
| Published online | 15 mars 2006 | |
Activité dominante négative des protéines p53 mutées
Dominant negative activity of mutated p53 proteins
1
Service de Génétique, Département de Pédiatrie, Faculté de médecine et des sciences de la santé, Université de Sherbrooke, 3001, 12e avenue Nord, Sherbrooke (Québec), J1H 5N4, Canada
2
Service de Cytogénétique, Département de Pathologie, Hôpital Sainte-Justine, 3175, chemin de la Côte-Sainte-Catherine, Montréal (Québec), H3T 1C5, Canada
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
La protéine p53 dispose d’une fonction activatrice de l’expression de nombreux gènes cibles. Le rôle de facteur de transcription joué par la protéine p53 nécessite la formation d’une structure homotétramérique. Les résultats de certaines expérimentations montrent que les monomères p53 mutés ont la capacité de se lier à des monomères p53 sauvages pour constituer des complexes hétérotétramériques. La présence de monomères p53 mutés au sein de ces complexes hétérotétramériques peut avoir pour conséquence immédiate une inactivation des monomères sauvages. Cette capacité de liaison et d’inactivation des p53 mutées à l’égard des p53 sauvages est qualifiée d’« effet dominant négatif ». Plusieurs facteurs enrôlés dans cette activité dominante négative ont été identifiés. La compréhension des fonctions moléculaires complexes qui régissent cette activité constitue un des aspects importants qui permettrait de mieux discerner les mécanismes biologiques en jeu dans la cancérogenèse. Le but de cet article est de mettre en lumière des aspects jusqu’à présent occultés de l’activité dominante négative des protéines p53 mutées. De plus, nous allons souligner comment cette activité contribue à la cancérogenèse induite par les rayons ultraviolets.
Abstract
Tumor suppressor gene inactivation as proposed by the Knudson model implies a sequential inactivation of two alleles of a gene. For example, the first allele is inactivated by a missense mutation, and the second one is inactivated by a deletion or insertion. The alteration of the p53 tumor suppressor gene is far to correspond only to this model. In the great majority of cancers, the mutated allele of p53 coexists with the normal allele. It is well known that the transcriptional activity is one of the most important functions of p53. The p53 protein is active as a tetramer (this complex activates the expression of targeted genes by binding to its consensus DNA sequence called the p53 response element). Experimental evidence shows that wild-type p53 interacts with mutant proteins to form heterotetramers. In association with wild-type proteins, mutant proteins drive the wild-type subunits into a mutant conformation. This association leads to a loss of trans-activating function. The capacity of mutant subunits to form heterotetramers with wild-type subunits and to commit them into a mutant conformation is called « dominant negative effect ». Many p53 mutant proteins possess this dominant negative activity. Recently, several factors, which are implicated in the control of the dominant negative activity of p53 mutants, have been identified. The elucidation of these complex molecular functions, which are implicated in the dominant negative activity of the p53 mutated protein represents an important aspect in the comprehension of the biological mechanisms involved in carcinogenesis.
© 2006 médecine/sciences - Inserm / SRMS
Parmi les multiples fonctions biologiques du gène suppresseur de tumeur p53 (Figure 1A), on retiendra celle de régulateur de l’expression de gènes cruciaux du cycle cellulaire, de la réparation des altérations de l’ADN génomique, de l’angiogenèse, de la sénescence ou encore celle de la mort cellulaire programmée. Dans certaines conditions de dommages à l’ADN ou de stress cellulaires (hypoxie, dépolymérisation du fuseau mitotique…), la protéine p53 (Figure 1B) est séquestrée dans le compartiment nucléaire. Cette stabilisation nucléaire conduit à une accumulation de la protéine p53 et à une activation de ses fonctions transcriptionnelles. L’activité de p53 repose sur l’importance du pouvoir transactivateur qu’elle exerce sur un grand nombre de gènes tels que p21, MDM2, GADD45, BAX, XPC, XPE et 14-3-3σ [1]. Son activité transcriptionnelle a des conséquences diverses qui vont de l’arrêt de la croissance cellulaire à l’induction apoptotique. Ainsi, le rôle de p53 durant la transcription revêt une importance capitale dans la carcinogenèse et la progression néoplasique. La fonction onco-suppressive de p53 implique sa fixation spécifique à l’ADN. Elle joue un rôle crucial dans les processus de réparation des dommages à l’ADN. Dans certaines cellules cancéreuses où la protéine p53 est inactive, le point de contrôle G1/S du cycle cellulaire est défectueux, entraînant une croissance cellulaire anarchique [2].
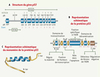 |
Figure 1. Structure du gène et de la protéine p53 humains. A. Le gène codant la protéine p53 humaine est localisé sur le bras court du chromosome 17, plus précisément sur la bande chromosomique 17p13.1. Il est composé de 11 exons et s’étend sur une longueur de 20 kilobases (kb). Il dispose d’une séquence intronique de 10 kb située entre la première et la deuxième séquence exonique. Cinq régions hautement conservées au cours de l’évolution sont localisées entre le second et le huitième exon. Il est généralement admis que le domaine central de la protéine p53 est codé par les exons 5, 6, 7, 8. B. La protéine p53 est une phosphoprotéine de 393 acides aminés. C. La structure cristallographique du monomère de la protéine p53 a été déterminée par Cho et al. [6]. Les principaux domaines de la protéine p53 sont : (1) un domaine aminoterminal (résidus 1-42) nécessaire à l’interaction avec les composantes de l’appareil transcriptionnel ; (2) une région riche en résidus proline (63-97) intervenant au cours de l’apoptose ; (3) un domaine central hydrophobe (102-292) dont la structure tridimensionnelle permet la fixation spécifique à l’ADN au sein duquel convergent la majorité des mutations inactivatrices en cause dans divers cancers humains ; (4) un domaine de tétramérisation (323-356) ; (5) un domaine carboxyterminal (363-393) intervenant dans la régulation négative de p53. |
L’inactivation d’un gène suppresseur de tumeur, telle que l’expose le modèle de Knudson, implique l’inactivation séquentielle des deux allèles. Le premier allèle serait inactivé par une mutation ponctuelle tandis que le second le serait par une délétion ou une insertion. L’altération du gène p53 est loin de répondre à ce modèle et obéit plutôt à une « logique inactivatrice » qui lui est particulière. La protéine p53 peut être inactivée par de nombreux mécanismes, notamment par l’un d’entre eux : l’effet de dominance négative des p53 mutées. Il s’agit de la capacité que possèdent les monomères p53 mutés de se lier et d’inactiver les monomères p53 sauvages (Figure 1C). Cet effet constitue l’un des principaux phénomènes qui participe à l’inactivation de p53 d’où son importance dans les cellules tumorales hétérozygotes pour le gène p53 (la majorité des cancers humains sont constitués de cellules coexprimant les allèles tant mutés que sauvages du gène p53). Au sein de ces cellules, l’effet dominant négatif peut devenir l’élément déclencheur de l’inactivation de la protéine issue de la copie sauvage de l’allèle de p53. À ce titre, le rôle de p53 dans la carcinogenèse cutanée constitue un exemple éloquent pour illustrer ce phénomène. La peau possède un mécanisme induit par p53 qui rétablit les dommages subis par l’ADN et lui permet d’éviter la formation de cellules transformées (kératinocytes p53−/−) (Figure 2). Cette fonction a valu à p53 le qualificatif de « gardien du tissu » [3].
 |
Figure 2. Modèle de Ziegler. Lors d’une exposition au soleil, les rayonnements ultraviolets de type B (UVB) peuvent causer des dommages à l’ADN (photoproduits) des cellules épidermiques, notamment les kératinocytes (voie 1). Ces cellules réparent la grande majorité des dommages, mais certaines peuvent acquérir une mutation ponctuelle C−T sur un des allèles du gène p53 (p53+/−) (voie 2) [3]. Lorsqu’il y a trop de dommages, la cellule évolue vers l’apoptose. Lors d’une exposition ultérieure, la protéine p53 est mobilisée pour procéder à la réparation des dommages à l’ADN. Les cellules normales (p53+/+) avec un grand nombre de dommages à réparer vont emprunter la voie de l’apoptose déclenchée par p53 (voie 3). Les cellules apoptotiques correspondent aux cellules « coup de soleil » (sunburn cells). Les kératinocytes p53+/− réparent les photoproduits moins efficacement et amorcent plus difficilement le processus apoptotique à cause de l’activité dominante négative des protéines p53 mutées (voie 4). Ces kératinocytes p53+/− possèdent alors une plus grande susceptibilité d’évoluer vers une cellule p53−/− (voie 5). Cette cellule p53−/− est une cellule transformée, elle peut alors donner naissance à un clone (voie 6) évoluant vers la kératose actinique précancéreuse, une étape préalable à l’émergence de carcinomes cutanés. L’activité dominante négative des protéines p53 mutées peut être enrôlée dans le mécanisme de résistance à l’apoptose et dans la diminution de l’efficacité de réparation au sein des kératinocytes p53+/−. |
Causes et conséquences de l’altération de l’expression de p53
L’altération du gène p53 se retrouve dans plus de la moitié des cancers humains [4] et son domaine central (102-292) regroupe à lui seul 90 % des mutations. L’inactivation de p53 est souvent causée par des mutations ponctuelles faux-sens de certains codons clés [4], particulièrement les codons 175, 248 et 273. Ces derniers sont considérés comme les codons les plus fréquemment mutés de la séquence génique de p53 et sont en cause dans l’émergence de nombreux cancers [5].
Parmi les divers mécanismes d’inactivation du gène p53 et de sa protéine qui ont été décrits, précisément dans le sens d’une perte de leur fonction, nous en évoquerons six qui nous paraissent les plus importants :
l’inactivation du gène p53 par délétion d’un ou de ses deux allèles qui réduit ou inhibe la formation du tétramère et conduit à une diminution de l’expression des gènes cibles ;
l’inactivation de la protéine p53 par :
mutations non-sens et mutations au sein du site d’épissage produisant des protéines tronquées incapables de s’oligomériser ;
mutations faux-sens des acides aminés du domaine central. Ces acides aminés substitués peuvent se répartir en deux groupes : un premier groupe comprenant les résidus engagés dans le maintien de la structure tridimensionnelle du domaine de liaison à l’ADN et un second groupe comprenant les résidus en contact direct avec l’ADN [6]. Ces deux catégories de mutations contribuent de façon directe à la génération de protéines pour lesquelles la capacité de liaison à l’ADN est abolie de façon partielle, voire totale ;
sa liaison avec certaines protéines virales telles que la protéine E6 du papillomavirus humain (HPV16 et 18) et l’antigène T du virus simien 40 (SV40) [7] ;
sa voie de signalisation qui peut être perturbée par altération de l’expression du gène MDM2 (double-minutes) qui est surexprimé et/ou amplifié [8] ;
défaillance des mécanismes de sa translocation conduisant de façon concomitante à sa séquestration cytoplasmique anormale et à son exclusion nucléaire [9].
Des formes particulières de mutations de p53 peuvent être à l’origine de mutants « gain de fonction ». La présence de ces mutants au sein des cellules engendre un phénotype nouveau. Ces dernières acquièrent de nouvelles fonctions normalement absentes chez les cellules avec la protéine p53 sauvage. Ces mutants « gain de fonction » disposent d’une activité dominante positive et peuvent prendre part à des processus oncogéniques [10]. On peut présenter comme exemple de cette activité dominante positive qui provient des mutations faux-sens, les protéines p53 mutées. Celles-ci peuvent en effet stimuler l’expression du gène MDR-1 (gène de résistance multiple aux drogues) laquelle est physiologiquement inhibée par la protéine p53 sauvage [11]. De récentes études in vivo faites sur des modèles murins ont prouvé avec clarté la validation du concept gain of fonction. Ces travaux montrent que sous certaines conditions physiologiques, l’expression du gène p53, porteur de mutations ponctuelles faux-sens, va au-delà de la simple perte de fonction de p53 et qu’elle augmente considérablement le potentiel oncogénique des cellules tumorales [12].
Les protéines p53 mutées peuvent également manifester un autre type d’activité dominante, plus communément appelée activité dominante négative. Celle-ci se manifeste par la capacité que possède la p53 mutée de se lier à p53 sauvage et à l’inactiver. Normalement, la p53 est active sous la forme d’un complexe homotétramérique (Figure 3 A et B) [13]. Cependant, certaines études montrent que la protéine p53 sauvage forme des complexes hétérotétramériques avec les protéines p53 mutées [14]. Ces dernières peuvent adopter deux conformations spatiales, l’une mutée et l’autre native. L’association des protéines p53 mutées avec les protéines p53 sauvages au sein du complexe hétérotétramérique entraîne les sous-unités p53 sauvages à adopter une conformation spatiale mutante [15]. Cette nouvelle conformation tridimensionnelle constitue un des mécanismes de l’activité dominante négative des protéines p53 mutées.
 |
Figure 3. A. Vue transversale d’un complexe tétramérique constitué de quatre monomères de la protéine p53. Chaque monomère est représenté par une couleur différente. Ce complexe tétramérique est assis sur son site de liaison spécifique à l’ADN, appelé élément de réponse de p53. B. Vue sagittale du complexe tétramérique sur son site spécifique de liaison à l’ADN. |
Dominants négatifs et facteurs de régulation
L’inactivation de p53 sauvage par des mutants dominants négatifs est tributaire de plusieurs facteurs, notamment l’environnement cellulaire [16], la stabilité protéique, le ratio sous-unités p53 sauvages/sous-unités mutantes au sein du complexe hétérotétramérique, la conformation spatiale adoptée par les protéines mutantes, la fonctionnalité du domaine de tétramérisation [17] et la nature du stimulus induisant p53 [18].
Chaque type cellulaire possède ses propres caractéristiques, par exemple des facteurs intrinsèques et des voies de signalisation spécifiques. L’environnement et la nature des cellules influencent en grande partie l’effet dominant négatif des p53 mutées [16].
Des expériences de cotraduction ont montré que l’inhibition de l’activité des sous-unités p53 sauvages était dépendante de la dose du mutant Arg273Leu [17]. Il apparaît donc, qu’une augmentation du nombre de sous-unités mutantes conduit à une inhibition de l’activité de la protéine p53 sauvage. Ce phénomène souligne l’importance de ce ratio quant à la détermination de la part occupée par l’activité dominante négative des protéines p53 mutées (Figure 4A).
 |
Figure 4. A. Rapport des sous-unités sauvages/mutées. Complexe tétramérique avec un ratio de trois monomères de la protéine p53 mutée (rouge) associés à un monomère sauvage (vert). Ce complexe tétramérique est assis sur son site spécifique de liaison à l’ADN. B. Conformations spatiales mutante et sauvage de la protéine p53. Illustration de deux monomères de la protéine p53 du complexe tétramérique p53 (les deux autres monomères ont été supprimés pour une meilleure compréhension). Le monomère p53 représenté en jaune adopte une conformation spatiale native. Le monomère p53 de couleur bleue présente une conformation spatiale mutante. Ces deux monomères sont assis sur leur site de liaison spécifique à l’ADN. Les flèches rouges indiquent le siège de la mutation, localisée au sein du domaine de liaison à l’ADN. C. Localisation des mutations sur les monomères de la protéine p53. Illustration de deux monomères du complexe tétramérique de p53 (les deux autres monomères ont été supprimés pour une meilleure compréhension). Sur le monomère p53 (bleu) est indiqué en rouge le siège de la mutation au sein du domaine de liaison à l’ADN. Sur le monomère p53 (violet) est indiquée en rouge la mutation au sein du domaine de tétramérisation. Ces deux monomères sont assis sur leur site de liaison spécifique à l’ADN. |
L’analyse de la structure tridimensionnelle des p53 mutées (Figure 4B) a permis de montrer que le mutant Arg175His présente d’importantes modifications conformationnelles faisant de lui un mutant structural [6]. En se liant aux sous-unités sauvages de p53, ce mutant les conduit à adopter une conformation spatiale mutante qui rend le complexe tétramérique incapable de se lier spécifiquement à sa séquence d’ADN. Cette restriction est imputable à l’activité dominante négative. Par opposition au mutant Arg175His, les mutants Arg273His, Arg273Leu et Arg248Trp possèdent une conformation spatiale native, à tout le moins une conformation qui se rapprocherait de celle de la protéine p53 sauvage [6].
Les mutations situées dans le domaine de tétramérisation ne représentent qu’un faible pourcentage de l’ensemble des mutations décrites. Cependant, toute mutation de cette sorte conduit les sous-unités mutées à perdre leur capacité de liaison aux sous-unités sauvages, induisant une perte de la capacité à former les complexes hétérotétramériques nécessaires pour exercer leur effet dominant négatif [19]. Le rôle du domaine de tétramérisation fonctionnel apparaît donc indispensable à l’activité dominante négative des protéines p53 mutées (Figure 4C).
Le traitement par les inhibiteurs de formation du fuseau mitotique (nocodazole et colcémide) des clones mutants 175His et 143Ala n’empêche pas leur progression dans le cycle cellulaire. En revanche, la lignée mutante 273His montre un arrêt post-métaphasique en G1 (arrêt de la reduplication de l’ADN, stade 4N ADN). Le mutant 273His peut se comporter comme la lignée sauvage LoVo (p53+/+) après un traitement par les inhibiteurs de formation du fuseau mitotique [18]. Le comportement de ce mutant s’expliquerait par la conformation native qu’il adopte. Pocard et al. ont étudié la réponse de p53 et p21 aux radiations ionisantes utilisées comme stimulus sur ces mêmes lignées cellulaires [20]. Ils ont constaté que les clones mutants 175His et 273His continuent leur progression dans le cycle cellulaire ; en revanche, ils ont observé que la lignée mutante 143Ala montre un arrêt post-métaphasique en G1. Les résultats de ces deux études suggèrent que la nature du stimulus pourrait être un facteur qui impose l’activité dominante négative des protéines p53 mutées.
Caractéristiques des mutants les plus fréquents de la protéine p53
Lorsque la composante majoritaire au sein du complexe tétramérique est la sous-unité mutée p53-273His, la conformation spatiale adoptée est alors native. Ce complexe peut avoir une activité transcriptionnelle résiduelle qui induirait l’expression de la protéine p21 [17]. Cette dernière induit l’arrêt du cycle cellulaire au stade G1 tétraploïde après traitement avec les inhibiteurs de formation du fuseau mitotique [18]. En revanche, cette lignée traitée aux radiations ionisantes ne montre pas d’arrêt en phase G1 tétraploïde [20]. Cette différence de réponse pourrait être liée à la nature du stimulus. Cependant, les mécanismes moléculaires qui sous-tendent ces réponses demeurent inconnus.
Le mutant Arg273Leu adopte une conformation spatiale native mais ne possède pas d’activité transcriptionnelle. Il est possible que les sous-unités p53 mutées au sein de ce complexe empêchent les sous-unités sauvages de se lier avec leurs éléments de réponse [21]. Les mécanismes moléculaires en jeu dans ce phénomène ne sont pas connus.
Le mutant Arg175His a une conformation spatiale mutante qui empêcherait sa fixation à son site de liaison à l’ADN. La conséquence immédiate est une absence totale d’activité transcriptionnelle [6]. C’est le cas d’une grande majorité des mutants p53 qui ne possèdent pas d’activité transcriptionnelle [22].
Le mutant Arg248Trp, malgré sa conformation spatiale native, ne peut pas se lier à son site de liaison à l’ADN. Ce phénomène expliquerait probablement l’absence d’activité transcriptionnelle [6]. La raison de ce comportement demeure inconnue.
Le mutant Val143Ala a une conformation spatiale mutante. Il présente un arrêt au stade G1 tétraploïde consécutif à un traitement par radiations ionisantes [20]. Cependant, les cellules porteuses de cette mutation continuent leurs progressions à travers le cycle cellulaire après un traitement par les inhibiteurs de formation du fuseau mitotique [18].
Nous voyons par ces exemples que le comportement de ces cinq mutants apparaît parfois en contradiction avec les résultats attendus. En effet, plusieurs points demeurent encore inexpliqués et exigent la compréhension d’autres mécanismes fonctionnels.
Après traitement des cellules par les inhibiteurs de formation du fuseau mitotique, le point de contrôle post-métaphasique semble dépendre de p53 sauvage, mais pas de l’activité transcriptionnelle de p53 mutée [23]. Cette étude permet de conclure à une perte de l’activité transcriptionnelle de p53 mutée qui continuerait cependant à assurer sa fonction de régulateur clé du cycle cellulaire. Par conséquent, d’autres mécanismes seraient à considérer. Les fonctions de la protéine p53 mutée ne semblent donc pas totalement abolies, mais seulement diminuées. La voie de signalisation p53/p21/pRb est perturbée, soulignant que les éléments en aval ou en parallèle à pRb seraient régulés par un signal indépendant de la transcription de p53. Ce signal, probablement transmis par le domaine p53 riche en proline, n’est pas nécessaire à l’activité transcriptionnelle de p53 [24]. Il est en revanche indispensable à l’arrêt de croissance en réponse à certains stimulus [25]. Il est également requis pour induire l’apoptose [24]. Par ailleurs, il a été montré que les résidus 73-143 de ce domaine interagissent directement avec le complexe hétérodimérique E2F/DP1 (famille de facteurs de transcription), inhibant son activité transcriptionnelle [26]. La compréhension des fonctions propres à ce domaine et sa portée dans l’arrêt post-métaphasique induit par les inhibiteurs du fuseau mitotique constituerait une étape importante dans la compréhension du rôle de p53 au cours du cycle cellulaire et au moment de la synthèse de l’ADN. L’ensemble de ces données est résumé dans le Tableau I.
Quelques caractéristiques de cinq mutants des plus fréquents de la protéine p53. (-) : non déterminé
Il est important de souligner l’existence d’activités dominantes négatives des protéines p53 mutées qui agissent indépendamment de la transcription et de l’expression de certains gènes. Dans leurs travaux, M. Mihara et al. [27] décrivent une activité dominante négative de mutants de p53 qui agissent par des mécanismes associant une localisation cytosolique et une interaction avec la mitochondrie [27]. Il est connu que la liaison de la protéine p53 sauvage avec les protéines BclXL et Bcl2 provoque la perméabilisation de la membrane mitochondriale externe. La perméabilisation de la membrane mitochondriale externe induit la libération du cytochrome C de la mitochondrie (espace intermembranaire) vers le cytosol, ce qui a pour effet le déclenchement de l’apoptose. Les cellules tumorales des mutants p53-273His et p53-175His sont incapables de former des complexes avec les protéines BclXL et Bcl2. Cependant, il y a un démarrage du processus de l’apoptose par l’intermédiaire de la libération du cytochrome C. Ces données suggèrent que ces mutants p53 possèdent une activité dominante négative indépendante de la transcription, associant une localisation cytosolique des protéines p53 mutées et leurs interactions avec la mitochondrie [27].
Intérêt thérapeutique
De multiples stratégies sont actuellement utilisées pour étudier p53 et ses voies de signalisations en œuvre dans la thérapeutique anticancéreuse. Dans la majorité des tumeurs, on constate une stabilisation de p53 mutée associée à son accumulation nucléaire, avec pour conséquence directe l’inhibition de la voie de dégradation dépendante de MDM2 [28]. Le niveau élevé de p53 mutée au sein des cellules cancéreuses, par rapport aux cellules saines, fournit une piste intéressante pour l’éclosion d’agents anti-prolifératifs. L’une des pistes de recherche que privilégie la compagnie ONYX Pharmaceuticals illustre clairement l’intérêt que suscite l’activité dominante négative des protéines p53 [29]. Les scientifiques de cette compagnie ont ciblé les cellules dont le taux de p53 mutées est élevé, associant ou pas des p53 sauvages, en utilisant l’adénovirus hybride ONYX-015. Cet adénovirus a été produit pour permettre une élimination ciblée restreinte aux cellules exprimant les protéines p53 mutées. Les cellules qui expriment la forme sauvage de la protéine ne sont pas affectées par la présence de ce virus. L’adénovirus qui se lie normalement à p53 est inactivé par une mutation, ce qui inhibe sa réplication dans les cellules contenant des protéines p53 fonctionnelles. En revanche, son potentiel réplicatif est conservé au sein des cellules cancéreuses ayant un gène p53 muté [30]. Les essais précliniques et cliniques sont prometteurs en vue de l’utilisation de ce virus pour le traitement des cancers de la tête et du cou. Toutefois, la spécificité exclusive de ce virus aux cellules qui expriment la forme mutée de p53 n’est pas encore prouvée d’une manière définitive [29].
Conclusions
Il est admis que la majorité des cancers humains sont constitués de cellules qui coexpriment l’allèle muté et l’allèle sauvage du gène p53. L’effet dominant négatif, qui se matérialise par la liaison et l’inactivation de la protéine p53 sauvage par la protéine p53 mutée, a en général pour conséquence la perte de la fonction p53. Selon le modèle de Knudson, la perte de la fonction normale d’un gène suppresseur de tumeur exige l’apparition successive de deux événements (mutation/délétion-insertion). Dans le cas particulier du gène p53, une mutation sur un allèle, par l’effet dominant négatif, va représenter à la fois le premier et le second événement du modèle de Knudson. L’inactivation de p53 peut survenir à différents stades de la carcinogenèse. La première mutation d’un allèle du gène p53 peut, par conséquent, contribuer au démarrage, à la promotion et à la progression du cancer et ce, en fonction du type cellulaire dans lequel la mutation est survenue. Dans cette perspective, de nouvelles stratégies thérapeutiques anticancéreuses ciblant p53 ou bénéficiant de la perte de son activité, pourraient être développées dans un proche avenir. Ces approches permettraient de concevoir de nouveaux outils diagnostiques et thérapeutiques.
Article reçu le 3 mai 2005, accepté le 29 septembre 2005.
Références
- Blagosklonny MV. P53 from complexity to simplicity : mutant p53 stabilization, gain-of-function, and dominant-negative effect. FASEB J 2000; 14 : 1901–7. [Google Scholar]
- Sherr CJ, Roberts JM. CDK inhibitors : positive and negative regulators of G1-phase progression. Genes Dev 1999; 12 : 1501–12. [Google Scholar]
- Ziegler A, Jonason AS, Leffell DJ, et al. Sunburn and p53 in the onset of skin cancer. Nature 1994; 372 : 773–6. [Google Scholar]
- Hollstein M, Sidransky D, Vogelstein B, Harris CC. P53 mutations in human cancers. Science 1991; 253 : 49–53. [Google Scholar]
- Hainaut P, Hollstein M. P53 and human cancer : the first ten thousand mutations. Adv Cancer Res 2000; 77 : 81–137. [Google Scholar]
- Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science 1994; 265 : 346–55. [Google Scholar]
- Munger K, Scheffner M, Huibregtse JM, Howley PM. Interactions of HPV E6 and E7 oncoproteins with tumour suppressor gene products. Cancer Surv 1992; 12 : 197–217. [Google Scholar]
- Oliner JD, Pietenpol JA, Thiagalingam S, et al. Oncoprotein MDM2 conceals the activation domain of tumour suppressor 53. Nature 1993; 29 : 857–60. [Google Scholar]
- Moll UM, Ostermeyer AG, Haladay R, et al. Cytoplasmic sequestration of wild-type p53 protein impairs the G1 checkpoint after DNA damage. Mol Cell Biol 1996; 3 : 1126–37. [Google Scholar]
- Dittmer D, Pati S, Zambetti G, et al. Gain of function mutations in p53. Nat Genet1993; 4 : 42–6. [Google Scholar]
- Lin J, Teresky AK, Levine AJ. Two critical hydrophobic amino acids in the N-terminal domain of the p53 protein are required for the gain of function phenotypes of human p53 mutants. Oncogene1995; 12 : 2387–90. [Google Scholar]
- Olive KP, Tuveson DA, Ruhe ZC, et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 2004; 119 : 847–60. [Google Scholar]
- Friedman PN, Chen X, Bargonetti J, Prives C. The p53 protein is an unusually shaped tetramer that binds directly to DNA. Proc Natl Acad Sci USA 1993; 90 : 3319–23. [Google Scholar]
- Brachmann RK, Vidal M, Boeke JD. Dominant-negative p53 mutations selected in yeast hit cancer hot spots. Proc Natl Acad Sci USA 1996; 9 : 4091–5. [Google Scholar]
- Milner J, Medcalf EA. Cotranslation of activated mutant p53 with wild type drives the wild-type p53 protein into the mutant conformation. Cell 1991; 65 : 765–7. [Google Scholar]
- Forrester K, Lupold SE, Ott VL, et al. Effects of p53 mutants on wild-type p53-mediated transactivation are cell type dependent. Oncogene 1995; 11 : 2103–11. [Google Scholar]
- Chene P. In vitro analysis of the dominant negative effect of p53 mutants. J Mol Biol 1998; 281 : 205–9. [Google Scholar]
- Dridi W, Fetni R, Lavoie J, et al. The dominant negative effect of p53 mutants and p21 induction in tetraploid G1 arrest depends on the type of p53 mutation and the nature of the stimulus. Cancer Genet CytoGenet 2003; 143 : 39–49. [Google Scholar]
- Chene P, Bechter E. P53 mutants without a functional tetramerisation domain are not oncogenic. J Mol Biol 1999; 5 : 1269–74. [Google Scholar]
- Pocard M, Chevillard S, Villaudy J, et al. Different p53 mutations produce distinct effects on the ability of colon carcinoma cells to become blocked at the G1/S boundary after irradiation. Oncogene 1996; 12 : 875–82. [Google Scholar]
- Willis A, Jung EJ, Wakefield T, Chen X. Mutant p53 exerts a dominant negative effect by preventing wild-type p53 from binding to the promoter of its target genes. Oncogene 2004; 23 : 2330–8. [Google Scholar]
- Monti P, Campomenosi P, Ciribilli Y. Tumour p53 mutations exhibit promoter selective dominance over wild type p53. Oncogene 2002; 21 : 1641–8. [Google Scholar]
- Notterman D, Young S, Wainger B, Levine AJ. Prevention of mammalian DNA reduplication, following the release from the mitotic spindle checkpoint, requires p53 protein, but not p53-mediated transcriptional activity. Oncogene 1998; 26 : 2743–51. [Google Scholar]
- Walker KK, Levine AJ. Identification of a novel p53 functional domain that is necessary for efficient growth suppression. Proc Natl Acad Sci USA 1996; 93 : 15335–40. [Google Scholar]
- Del Sal G, Ruaro EM, Utrera R, et al. Gas1-induced growth suppression requires a transactivation-independent p53 function. Mol Cell Biol 1995; 15 : 7152–60. [Google Scholar]
- Sorensen TS, Girling R, Lee CW, et al. Functional interaction between DP-1 and p53. Mol Cell Biol 1996; 16 : 5888–95. [Google Scholar]
- Mihara M, Erster S, Zaika A, et al. p53 has a direct apoptogenic role at the mitochondria. Mol Cell 2003; 11 : 577–90. [Google Scholar]
- Whitesell L, Sutphin P, An WG, et al. Geldanamycin-stimulated destabilization of mutated p53 is mediated by the proteasome in vivo. Oncogene 1997; 23 : 2809–16. [Google Scholar]
- Lane DP. Killing tumor cells with viruses : a question of specificity. Nat Med 1998; 9 : 1012–3. [Google Scholar]
- Ganly I, Kirn D, Eckhardt G, et al. A phase I study of Onyx-015, an E1B attenuated adenovirus, administered intratumorally to patients with recurrent head and neck cancer. Clin Cancer Res 2000; 3 : 798–806. [Google Scholar]
Liste des tableaux
Quelques caractéristiques de cinq mutants des plus fréquents de la protéine p53. (-) : non déterminé
Liste des figures
|
Figure 1. Structure du gène et de la protéine p53 humains. A. Le gène codant la protéine p53 humaine est localisé sur le bras court du chromosome 17, plus précisément sur la bande chromosomique 17p13.1. Il est composé de 11 exons et s’étend sur une longueur de 20 kilobases (kb). Il dispose d’une séquence intronique de 10 kb située entre la première et la deuxième séquence exonique. Cinq régions hautement conservées au cours de l’évolution sont localisées entre le second et le huitième exon. Il est généralement admis que le domaine central de la protéine p53 est codé par les exons 5, 6, 7, 8. B. La protéine p53 est une phosphoprotéine de 393 acides aminés. C. La structure cristallographique du monomère de la protéine p53 a été déterminée par Cho et al. [6]. Les principaux domaines de la protéine p53 sont : (1) un domaine aminoterminal (résidus 1-42) nécessaire à l’interaction avec les composantes de l’appareil transcriptionnel ; (2) une région riche en résidus proline (63-97) intervenant au cours de l’apoptose ; (3) un domaine central hydrophobe (102-292) dont la structure tridimensionnelle permet la fixation spécifique à l’ADN au sein duquel convergent la majorité des mutations inactivatrices en cause dans divers cancers humains ; (4) un domaine de tétramérisation (323-356) ; (5) un domaine carboxyterminal (363-393) intervenant dans la régulation négative de p53. |
| Dans le texte | |
|
Figure 2. Modèle de Ziegler. Lors d’une exposition au soleil, les rayonnements ultraviolets de type B (UVB) peuvent causer des dommages à l’ADN (photoproduits) des cellules épidermiques, notamment les kératinocytes (voie 1). Ces cellules réparent la grande majorité des dommages, mais certaines peuvent acquérir une mutation ponctuelle C−T sur un des allèles du gène p53 (p53+/−) (voie 2) [3]. Lorsqu’il y a trop de dommages, la cellule évolue vers l’apoptose. Lors d’une exposition ultérieure, la protéine p53 est mobilisée pour procéder à la réparation des dommages à l’ADN. Les cellules normales (p53+/+) avec un grand nombre de dommages à réparer vont emprunter la voie de l’apoptose déclenchée par p53 (voie 3). Les cellules apoptotiques correspondent aux cellules « coup de soleil » (sunburn cells). Les kératinocytes p53+/− réparent les photoproduits moins efficacement et amorcent plus difficilement le processus apoptotique à cause de l’activité dominante négative des protéines p53 mutées (voie 4). Ces kératinocytes p53+/− possèdent alors une plus grande susceptibilité d’évoluer vers une cellule p53−/− (voie 5). Cette cellule p53−/− est une cellule transformée, elle peut alors donner naissance à un clone (voie 6) évoluant vers la kératose actinique précancéreuse, une étape préalable à l’émergence de carcinomes cutanés. L’activité dominante négative des protéines p53 mutées peut être enrôlée dans le mécanisme de résistance à l’apoptose et dans la diminution de l’efficacité de réparation au sein des kératinocytes p53+/−. |
| Dans le texte | |
|
Figure 3. A. Vue transversale d’un complexe tétramérique constitué de quatre monomères de la protéine p53. Chaque monomère est représenté par une couleur différente. Ce complexe tétramérique est assis sur son site de liaison spécifique à l’ADN, appelé élément de réponse de p53. B. Vue sagittale du complexe tétramérique sur son site spécifique de liaison à l’ADN. |
| Dans le texte | |
|
Figure 4. A. Rapport des sous-unités sauvages/mutées. Complexe tétramérique avec un ratio de trois monomères de la protéine p53 mutée (rouge) associés à un monomère sauvage (vert). Ce complexe tétramérique est assis sur son site spécifique de liaison à l’ADN. B. Conformations spatiales mutante et sauvage de la protéine p53. Illustration de deux monomères de la protéine p53 du complexe tétramérique p53 (les deux autres monomères ont été supprimés pour une meilleure compréhension). Le monomère p53 représenté en jaune adopte une conformation spatiale native. Le monomère p53 de couleur bleue présente une conformation spatiale mutante. Ces deux monomères sont assis sur leur site de liaison spécifique à l’ADN. Les flèches rouges indiquent le siège de la mutation, localisée au sein du domaine de liaison à l’ADN. C. Localisation des mutations sur les monomères de la protéine p53. Illustration de deux monomères du complexe tétramérique de p53 (les deux autres monomères ont été supprimés pour une meilleure compréhension). Sur le monomère p53 (bleu) est indiqué en rouge le siège de la mutation au sein du domaine de liaison à l’ADN. Sur le monomère p53 (violet) est indiquée en rouge la mutation au sein du domaine de tétramérisation. Ces deux monomères sont assis sur leur site de liaison spécifique à l’ADN. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.