")
")
| Issue |
Med Sci (Paris)
Volume 21, Décembre 2005
Métabolisme glucidolipidique et risque cardiovasculaire: Nouvelle approches
|
|
|---|---|---|
| Page(s) | 26 - 29 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/20052111s26 | |
| Published online | 15 novembre 2005 | |
Manifestations neurologiques de la maladie de Fabry
Neurological aspects of Fabry disease
1
Unité de Neurologie Générale, CHU Michallon, 38043 Grenoble Cedex 9, France
2
Service de Neurologie, CHU Montpied, 58, rue Montalembert, 63000 Clermont-Ferrand, France
3
Unité Fonctionnelle de Génétique Clinique, Hôpital Européen Georges Pompidou, 20, rue Leblanc, 75015 Paris, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
La maladie de Fabry (MF), sphingolipidose lysosomale liée au chromosome X, est responsable d’une atteinte multisystémique, continuum de sévérité clinique en fonction du niveau d’activité α-galactosidase A. L’atteinte du système nerveux périphérique, avec des crises douloureuses et des acroparesthésies chroniques, souvent révélatrice, est en rapport avec une atteinte des fibres nerveuses de petit calibre (fibres δ), ce qui est confirmé par la normalité de l’électroneuromyographie et l’élévation des seuils à la douleur ou aux stimulations thermiques. Les atteintes cochléo-vestibulaires et du système nerveux autonome sont plus tardives. Outre de rares méningites aseptiques, les atteintes centrales sont dues à des lésions vasculaires ischémiques survenant dans 25 % des cas etaugmentant avec l’âge. Elles intéressent préférentiellement le territoire vertébro-basilaire et leurphysiopathologie n’est pas encore élucidée. L’IRM encéphalique montre l’existence de nombreuseslésions silencieuses, augmentant avec l’âge, dans les territoires vascularisés par des artères perforantes de petit calibre ainsi que des calcifications des pulvinars secondaires à l’hyperperfusion cérébrale observée dans la MF. Celle-ci pourrait être due à la microangiopathie secondaire aux dépôts de glycosphingolipides. L’enzymothérapie substitutive semble pouvoir améliorer les anomalies du métabolisme régional encéphalique, mais aussi les manifestations douloureuses liées à l’atteintepériphérique.
Abstract
Fabry disease, a rare X-linked disorder with deficient activity of alpha-galactosidase A, leads to a multiple organ failure caused by a progressive accumulation of the substrat globotriasocylceramide in cells. Peripheral nerve involvement, neuropathic pain and chronic acroparesthesiae, are the most frequently reported signs and often revealing the disease. They are secondary to the small nerve fibres (fibres δ), that explained the normality of electroneuromyography. Cochleo-vestibular and autonomic nervous system involvement appears later in the illness, aseptic meningitis are rare. Cerebrovascular events (stroke, transient ischaemic attack) are reported in 25 % of patients, increasing with age. Affecting essentially the posterior circulation, their etiologies have to be clarified. MRI shows numerous silent lesions, increasing with age, mainly in small perforant arteries and pulvinar calcifications, due to an increase in cerebral perfusion with an impaired cerebral autoregulation, secondary to the glycosphingolipid storage in vascular endothelial cells. Enzyme replacement therapy could improve cerebral regional blood flow disturbances and painful neuropathy.
© 2005 médecine/sciences - Inserm / SRMS
La maladie de Fabry (MF) est une sphingolipidose lysosomale de transmission génétique liée au chromosome X, responsable d’une atteinte multisystémique, initiée par l’accumulation diffuse de glycosphingolipides, au niveau des lysosomes de nombreux types cellulaires. Tous les organes peuvent être atteints d’où la grande variation des manifestations cliniques, faisant de la MF un modèle d’affection multi-systémique. Alors que cette maladie génétique est connue depuis 1898, l’atteinte neurologique n’a été rapportée pour la première fois qu’en 1952 sur des données anatomopathologiques [1]. Les neurologues se doivent pourtant de la connaître puisque l’atteinte du système nerveux périphérique est fréquente et précoce dans la MF, rapidement complétée par une atteinte du système nerveux autonome et de l’appareil cochléo-vestibulaire tandis que l’atteinte centrale, dominée par les événements vasculaires cérébraux, prédominant curieusement sur la circulation vertébro-basilaire et intéressant aussi bien l’homme que la femme [2, 3] font avec l’insuffisance rénale et la cardiomyopathie hypertrophique tout le pronostic de l’affection après l’âge de 35 ans. L’efficacité de l’enzymothérapie substitutive par α-galactosidase A recombinante [4] pour la prévention des atteintes neurologiques centrales reste à préciser.
Atteinte du système nerveux périphérique
Les manifestations neurologiques périphériques, débutant dans l’enfance, sont dominées par les crises douloureuses aiguës et les acroparesthésies chroniques, signes par ailleurs le plus souvent rapportés par les patients (84 % des garçons et 79 % des filles) [5]. Il s’agit de brûlures ou de picotements des paumes des mains et des plantes des pieds, plus rarement de l’hémiface [6] pendant quelques minutes ou, de façon permanente, pendant plusieurs jours. Elles sont favorisées par la fatigue, l’exercice physique, le stress, les variations climatiques brutales [6] et parfois associées à une fièvre, un œdème des chevilles ou des arthralgies.
Des crises douloureuses aiguës fulgurantes à type de coup de poignard ou en décharges électriques [7] peuvent se surajouter aux acroparesthésies chroniques.
La thérapeutique repose sur les antiépileptiques, carbamazépine, diphényl-hydantoïne ou gabapentine [8]. L’efficacité exacte de la thérapie enzymatique substitutive sur la douleur reste à préciser [9].
L’atteinte des fibres nerveuses de petit calibre (fibres δ) est confirmée : (1) par le fait que ces patients ont un seuil élevé de sensibilité à la chaleur et encore plus au froid au niveau des extrémités [6, 10] ; (2) par la normalité de l’électroneuromyogramme [11]. Cette neuropathie des petites fibres permettrait d’expliquer les anomalies de réponse (diminution du tonus vasoconstricteur et augmentation de la vasodilatation périphérique) en cas d’ischémie [12].
La physiopathologie des manifestations douloureuses reste mal comprise. L’infiltration des ganglions de la racine dorsale de la moëlle et des gaines des fibroblastes périneuraux par des dépôts de Gb3 a été évoquée [13, 14]. Enfin, on ne peut pas écarter d’éventuelles anomalies de la vascularisation et l’occlusion des vasa-nervorum dans la genèse des crises douloureuses aiguës.
Atteinte des nerfs crâniens
L’atteinte des nerfs crâniens prédomine sur les nerfs oculomoteurs, volontiers retrouvée en cas de maladie cérébrovasculaire du fait de l’atteinte préférentielle de la circulation postérieure, mais d’autres nerfs crâniens (V, VII et XII) peuvent être touchés, parfois par compression par une artère vertébrale dolichoectasique [15].
L’atteinte cochléo-vestibulaire est dominée par la surdité, endocochléaire, rétrocochléaire, ou mixte, progressive, uni- ou bilatérale, et s’aggravant avec l’âge [16]. Des signes vestibulaires périphériques polymorphes, allant de simples sensations vertigineuses jusqu’au vertige vrai, parfois positionnel, sont fréquemment observés [16].
Atteinte du système nerveux autonome
L’atteinte du système nerveux autonome, objectivée par la suppression du réflexe cutané sympathique, est liée à l’accumulation de glycosphingolipides dans les ganglions spinaux et les ganglions sympathiques [13]. Elle se traduit par une hypotension orthostatique, un trouble de la motilité intestinale (diarrhées post-prandiales, crampes abdominales) et une diminution des sécrétions sudorales, salivaires et lacrymales [17].
Il faut noter l’accumulation de glycolipides dans les cellules du système nerveux autonome au niveau du tronc cérébral, dans les ganglions sympathiques, dans le tractus intermediolateralis de la moelle, dans les plexus sous-muqueux, dans les noyaux amygdaliens. L’atteinte du noyau hypothalamique pourrait expliquer l’anhidrose, l’intolérance aux températures extrêmes et la fièvre.
Atteinte du système nerveux central
Les complications cérébro-vasculaires, fréquentes au cours de la MF (20 à 30 % des patients) [2, 5], augmentent avec l’âge, survenant en moyenne entre 28 et 34 ans chez les hommes et entre 40 et 44 ans chez la femme. Elles peuvent être exceptionnellement révélatrice de la MF, y compris chez la femme [18] mais n’ont jamais été décrites avant l’âge de 12 ans [5]. Les récidives sont fréquentes, notées chez 76 % des hémizygotes et 55 % des hétérozygotes, en moyenne 6 ans après le premier événement [15], et pouvant progressivement induire un syndrome pseudo-bulbaire et/ou un syndrome démentiel.
Les accidents vasculaires cérébraux (AVC) sont, dans la majorité des cas, ischémiques, localisés dans le territoire vertébro-basilaire (67 % chez les hémizygotes, 60 % chez les hétérozygotes). Les symptômes sont dominés par les signes d’atteinte du territoire postérieur : hémiparésie (63 %), vertiges (39 %), diplopie (37 %), dysarthrie (30 %), nystagmus (26 %) et ataxie (21 %). Les accidents hémorragiques, conséquence d’un mauvais contrôle de l’hypertension artérielle sont plus rares [15].
La physiopathologie des complications cérébrovasculaires de la MF, ainsi que la localisation préférentielle du territoire artériel vertébro-basilaire, ne sont pas encore totalement élucidées [19]. On évoque plusieurs facteurs à l’origine de l’ischémie cérébrale : sténose progressive des petits vaisseaux secondaire à un dépôt de glycosphingolipides dans la paroi vasculaire, aggravée par un dysfonctionnement endothélial, absence de fonctionnalité des voies vasodilatatrices [20], remodelage artériel [21], état pro-thrombotique [22], hyperaggrégabilité plaquettaire, modification de la régulation de la perfusion cérébrale liée à une dysautonomie [23], athérosclérose diffuse des gros troncs, embolie d’origine cardiaque sur une valvulopathie ou des troubles du rythme, hypertension artérielle d’origine rénale. Enfin, il faut envisager la fréquence des dolichoectasies des artères intracrâniennes, probablement en relation avec les dépôts de glycosphingolipides dans les cellules musculaires lisses de la paroi artérielle et pouvant potentiellement contribuer à la formation d’un thrombus du tronc artériel susceptible d’emboliser dans le lit d’aval, ou d’un étirement avec obstruction des vaisseaux naissant de l’artère dolichoectasique [15].
L’IRM cérébrale révèle des lésions corticales ou sous-corticales, sus- ou sous-tentorielles, des hyper-signaux de la substance blanche périventriculaire (Figure 1) et des dolichoectasies des vaisseaux vertébro-basilaires. Elle n’est normale que chez 25 % des patients hémizygotes et 40% des patientes hétérozygotes [15]. Une étude longitudinale d’une durée de 4 ans, réalisée chez 50 patients hémizygotes a permis de mettre en évidence des lésions cérébrales sur les séquences pondérées en T2 chez 55 % des patients [24], alors que 37,5 % seulement des patients avaient présenté un événement symptomatique, constitué ou transitoire. La majorité des premières lésions apparaissaient dans les territoires vascularisés par des artères perforantes de petit calibre, essentiellement au niveau de la substance blanche périventriculaire, des noyaux gris centraux et dans le tronc cérébral ou le cervelet. Il existait enfin une augmentation progressive de la charge lésionnelle à l’IRM avec l’âge, puisque tous les patients de plus de 54 ans présentaient des lésions cérébrales.
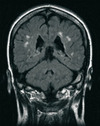 |
Figure 1. Maladie de Fabry : IRM encéphalique montrant des hypersignaux périventriculaires diffus de petite taille (lacunes) intéressant la substance blanche (coupe frontale, séquence FLAIR). |
Deux travaux ont permis de noter de fréquentes calcifications des pulvinars chez les patients hémizygotes [25, 26]. Cette anomalie, augmentant avec l’âge puisque présente chez plus de 30 % des patients de plus de 50 ans, pourrait être une caractéristique distinctive de la MF. Elle pourrait être secondaire à la vulnérabilité des pulvinars et des noyaux thalamiques adjacents par augmentation du débit sanguin cérébral (DSC) confirmé par des études de doppler trans-crânien [23]. Enfin, les travaux en spectroscopie ont permis de noter qu’il existait une atteinte cérébrale diffuse, notamment des régions sous-corticales, avec diminution du pic de N-acétyl aspartate, même en l’absence de lésions visibles en imagerie conventionnelle [27]. Cette souffrance neuronale pourrait être secondaire aux dépôts neuronaux de substrats [17] ou à un dysfonctionnement issu des modifications vasculaires [27]. Il existe ainsi une diminution du métabolisme régional du glucose et une augmentation du débit local au niveau de la substance blanche (SB) chez les patients atteints de MF, en caméra à positons [19]. Ainsi, l’augmentation du DSC local et la réactivité altérée des mécanismes de régulation artérielle seraient responsables d’une augmentation de la pression interstitielle locale et d’une vulnérabilité métabolique de la substance blanche (SB) [19]. Si l’augmentation du DSC est due à la microangiopathie secondaire aux dépôts de glycosphingolipides ou aux dysfonctionnements endothéliaux induits par les glycosphingolipides, la thérapeutique substitutive devrait pouvoir corriger l’augmentation du DSC régional, comme cela est observé dans l’artère sylvienne par doppler transcrânien et au niveau vertébro-basilaire par caméra à positons [28].
Traitement
Il n’y a pas de traitement spécifique des complications cérébrovasculaires de la MF. Il convient d’insister sur la prévention thromboembolique systématique par les antiagrégants plaquettaires [29] ou les anticoagulants en cas de risque cardio-embolique [29]. Le profil lipidique doit être contrôlé et une hyperhomocystéinémie corrigée par l’administration d’acide folique lorsqu’elle existe [30]. L’enzymothérapie substitutive, par α-galactosidase recombinante (1 mg/kg de poids d’agalsidase β administrée en perfusion intraveineuse chaque 15 jours), a permis la normalisation des taux plasmatiques de Gb3 [4, 31], la clairance du Gb3 des cellules endothéliales rénales [31] et d’autres types cellulaires du rein [32] et de la peau, et la stabilisation de la fonction rénale [4]. Dans une récente étude multicentrique de phase IV, contrôlée en double aveugle contre placebo, le nombre d’événements morbides rénaux, cardiaques et cérébro-vasculaires a été significativement réduit dans le groupe traité par agalsidase β (1 mg/kg) par rapport au groupe témoin. En revanche, il existe quelques observations d’accidents ischémiques transitoires survenant malgré la thérapie enzymatique substitutive. Il semble logique de considérer la thérapie enzymatique, par ailleurs bien tolérée, comme un traitement préventif à débuter le plus précocement possible afin de prévenir des lésions irréversibles [29].
Conclusion
Il apparaît clairement qu’un effort d’éducation médicale doit être accompli pour favoriser un diagnostic plus précoce pouvant conduire à l’instauration d’un traitement préventif. Sur le plan épidémiologique, des études systématiques de dépistage sur des populations de sujets adultes jeunes ayant présenté un AVC ischémique inexpliqué, pourraient permettre de mieux préciser la place exacte de la maladie de Fabry parmi les étiologies d’accidents vasculaires cérébraux.
Références
- Hornbostel H. Das angiokeratoma corporis diffusum universale mit kardio-vaso-renalem symptomenkomplex als neuartige thesaurismoseform. Helvetica Medica Acta 1952; 19 : 388–96. [Google Scholar]
- MacDermot KD, Holmes A, Miners AH. Anderson-Fabry disease : clinical manifestations and impact of the disease in a cohort of 98 hemizygous males.J Med Genet 2001; 38 : 750–60. [Google Scholar]
- Whybra C, Kampmann C, Willers I, et al. Anderson-Fabry disease: clinical manifestations of disease in female heterozygotes. J Inherit Metab Dis 2001; 24 : 715–24. [Google Scholar]
- Wilcox W, Banikazemi M, Guffon N, et al. Long-term safety and efficacity of enzyme replacement therapy Fabry disease. Am J Hum Genet 2004; 75 : 65–74. [Google Scholar]
- Mehta A, Ricci R, Widmer U, et al. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry outcome survey. Eur J Clin Invest 2004; 34 : 236–42. [Google Scholar]
- Valeriani M, Mariotti P, Le Pera D, et al. Functional assessment of A delta and C fibers in patients with Fabry’s disease. Muscle Nerve 2004; 30 : 708–13. [Google Scholar]
- Beck M, Whybra C, Wendrick K, et al. Fabry disease in children and adolescent. Contrib Nephrol 2001; 136 : 251–5. [Google Scholar]
- Filling-Katz MR, Merrick HF, Fink JK, et al. Carbamazepine in Fabry’s disease: effective analgesia with dose-dependent exacerbation of autonomic dysfunction. Neurology 1989; 39 : 598–600. [Google Scholar]
- Hilz MJ, Brys M, Marthol H, et al. Enzyme replacement therapy improves function of C-, Adelta-, and Abeta-nerve fibers in Fabry neuropathy. Neurology 2004; 62 : 1066–72. [Google Scholar]
- Luciano CA, Russell JW, Banerjee TK, et al. Physiological characterization of neuropathy in Fabry’s disease. Muscle Nerve 2002; 26 : 622–9. [Google Scholar]
- Gomes I, Nora DB, Becker J, et al. Nerve conduction studies, electromyography and sympathetic skin response in Fabry’s disease. J Neurol Sci 2003; 214 : 21–5. [Google Scholar]
- Lacomis D, Roeske-Anderson L, Mathie L. Neuropathy and Fabry’s disease. Muscle Nerve 2005; 31 : 102–7. [Google Scholar]
- Hozumi I, Nishizawa M, Ariga T, et al. Accumulation of glycosphingolipids in spinal and sympathetic ganglia of a symptomatic heterozygote of Fabry’s disease. J Neurol Sci 1989; 90 : 273–80. [Google Scholar]
- De Veber GA, Schwarting GA, et al. Fabry disease : immunocytochemical characterization of neuronal involvement. Ann Neurol 1992; 31 : 409–15. [Google Scholar]
- Mitsias P, Levine SR. Cerebrovascular complications of Fabry’s disease. Ann Neurol 1996; 40 : 8–17. [Google Scholar]
- Germain DP, Avan P, Chassaing A, et al. Patients affected with Fabry disease have an increased incidence of progressive hearing loss and sudden deafness: an investigation of twenty-two hemizygous male patients. BMC Med Genet 2002; 3 : 10. [Google Scholar]
- Cable WJ, Kolodny EH, Adams RD. Fabry disease : impaired autonomic function. Neurology 1982; 32 : 498–502. [Google Scholar]
- Giacomini PS, Shannon PT, Clarke JT, et al. Fabry’s disease presenting as stroke in a young female. Can J Neurol Sci 2004; 31 : 112–4. [Google Scholar]
- Moore DF, Altarescu G, Braker WC, et al. White matter lesion in Fabry disease occurs in prior selectively hypometabolic and hyperperfused brain regions. Brain Res Bull 2003; 62 : 231–40. [Google Scholar]
- Moore DF, Scott LTC, Gladwin MT, et al. Regional cerebral hyperperfusion and nitric oxide pathway dysregulation in Fabry disease. Reversal after enzyme replacement. Circulation 2001; 104 : 1506–12. [Google Scholar]
- Boutouyrie P, Laurent S, Laloux B, et al. Non- invasive evaluation of arterial involvement in patients affected with Fabry disease. J Med Genet 2001; 38 : 629–31. [Google Scholar]
- DeGraba T, Azhar S, Dignat-George F, et al. Profile of endothelial and leukocyte activation in Fabry patients. Ann Neurol 2000; 47 : 229–33. [Google Scholar]
- Hilz MJ, Kolodony EH, Brys M, et al. Reduced cerebral blood flow velocity and impaired cerebral autoregulation in patients with Fabry disease. J Neurol 2004; 251 : 564–70. [Google Scholar]
- Crutchfield KE, Patronas NJ, Dambrosia JM, et al. Quantitative analysis of cerebral vasculopathy in patients with Fabry disease. Neurology 1998; 50 : 1746–9. [Google Scholar]
- Moore DF, Ye F, Schiffmann R, et al. Increased signals intensity in the pulvinar on T1-weighted images: a pathognomonic MR imaging sign of Fabry disease. Am J Neuroradiol 2003; 62 : 231–40. [Google Scholar]
- Takanashi JI, Barkovich J, Dillon WP, et al. T1 hyperintensity in the pulvinar : key imaging feazture for diagnosis of Fabry disease. Am J Neuroradiol 2003; 24 : 916–21. [Google Scholar]
- Tedeschi G, Bonavita S, Banerjee TK, et al. Diffuse central neuronal involvement in Fabry disease: a proton MRS imaging study. Neurology 1999; 52 : 1663–7. [Google Scholar]
- Moore DF, Altarescu G, Ling GS, et al. Elevated cerebral blood flow velocities in Fabry disease with reversal after enzyme replacement. Stroke 2002; 33 : 525–31. [Google Scholar]
- Germain DP. La maladie de Fabry en 2004 : l’avènement de la thérapeutique. Rev Prat 2003; 53 : 2215–20. [Google Scholar]
- Demuth K, Germain DP. Endothelial markers and homocysteine in patients with classic Fabry disease. Acta Paediatr 2002; 91 : 57–61. [Google Scholar]
- Eng CM, Guffon N, Wilcox WR, et al. Safety and efficacy of recombinant human alpha-galactosidase replacement therapy in Fabry’s disease. N Engl J Med 2001; 345 : 9–16. [Google Scholar]
- Thurberg BL, Rennke H, Colvin RB, et al. Globotriaosylceramide in the Fabry kidney is cleared from multiple cell types after enzyme replacement therapy. Kidney Int 2002; 62 : 1933–46. [Google Scholar]
Liste des figures
|
Figure 1. Maladie de Fabry : IRM encéphalique montrant des hypersignaux périventriculaires diffus de petite taille (lacunes) intéressant la substance blanche (coupe frontale, séquence FLAIR). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.