")
")
| Issue |
Med Sci (Paris)
Volume 21, Décembre 2005
Métabolisme glucidolipidique et risque cardiovasculaire: Nouvelle approches
|
|
|---|---|---|
| Page(s) | 20 - 22 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/20052111s20 | |
| Published online | 15 novembre 2005 | |
Rein et maladie de Fabry : mécanismes de progression et de régression de la fibrose rénale vasculaire
Kidney and Fabry disease: mechanisms of renal vascular fibrosis
Inserm U.702, Hôpital Tenon, 4, rue de la Chine, 75020 Paris, France
AP-HP, Laboratoire de Physiologie, Faculté de Médecine Saint-Antoine, Université Pierre et Marie Curie, 27, rue de Chaligny, 75012 Paris, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Il est aujourd’hui admis que la progression de l’insuffisance rénale au cours de la maladie de Fabry est liée à l’apparition de lésions dans les vaisseaux intrarénaux. Dans cet article, nous aborderons les progrès récents faits dans la compréhension des mécanismes de progression et de régression de la fibrose vasculaire rénale. Ces travaux, réalisés dans des modèles expérimentaux animaux, établissent le rôle important joué par des agents profibrosants tels l’angiotensine II, l’endothéline et le TGFβ. Les traitements pharmacologiques bloquant ou antagonisant l'action des ces agents profibrosants ont pu bloquer la progression de la fibrose et ont rétabli partiellement, et même dans certains cas complètement, la fonction et la structure rénale. Nous espérons que, dans un avenir proche, il sera possible de proposer aux patients des traitements efficaces contre le développement de la fibrose rénale associée aux différentes néphropathies.
Abstract
In Fabry disease, the critical step for progression towards renal failure is the appearance of ischemic lesions within renal vesserls. Recent studies in experimental models identified angiotensin II, endothelin and growth factors as key players in the mechanisms of nephroangiosclerosis. In addition, they showed that renal fibrosis is a reversible process providing thus, hope for efficient treatment(s) against chronic renal failure.
© 2005 médecine/sciences - Inserm / SRMS
Bien que le diagnostic histologique de la maladie de Fabry soit généralement fondé sur l’apparition des lésions de vacuolisation dans les podocytes, cette anomalie ne constitue pas l’étape déterminante vers l’insuffisance rénale chronique. La maladie de Fabry évolue vers une insuffisance rénale quand les cellules de l’environnement des podocytes telles que les cellules endothéliales, mésangiales et musculaires lisses des artérioles sont atteintes ; à partir de cette étape, des lésions cicatricielles, non spécifiques de la maladie de Fabry, caractéristiques d’une ischémie rénale se développent. Dans cet article, nous exposerons les résultats acquis ces dernières années par notre équipe sur les mécanismes de progression de l’insuffisance rénale dans des modèles expérimentaux d’ischémie rénale.
Développement de la fibrose rénale vasculaire
Historiquement, les facteurs hémodynamiques ont été les premiers décrits comme responsables de la progression de l’insuffisance rénale chronique. L’hypertension artérielle systémique, qu’elle soit à l’origine ou qu’elle résulte de l’insuffisance rénale chronique, accélère par elle-même le déclin de la fonction rénale, essentiellement par transmission de l’hypertension artérielle aux capillaires glomérulaires. La néphroangiosclérose est chez l’homme un exemple typique de néphropathie vasculaire au cours de laquelle les processus pro-fibrosants intéressent les microvaisseaux corticaux, les glomérules, puis l’interstitium rénal. L’effet néphroprotecteur des traitements antihypertenseurs est actuellement sans équivoque dans la plupart des néphropathies [1–4].
Les mécanismes de développement de la fibrose rénale au cours de la néphroangiosclérose, en dehors des anomalies hémodynamiques font intervenir les effets trophiques des peptides vasoactifs comme l’angiotensine II (Ang II). L’Ang II stimule la synthèse de protéines de la matrice extracellulaire, dont les différents collagènes, par les cellules mésangiales et les cellules musculaires lisses vasculaires. Grâce à l’utilisation de souris transgéniques, possédant le gène de la luciférase sous le contrôle du promoteur de la chaîne α2 du collagène de type I (col Iα2), nous avons pu analyser l’action in vivo de l’Ang II sur le gène du collagène I. En effet, chez ces souris, toute activation du promoteur se traduit par une augmentation de l’activité de la luciférase facilement décelable par sa luminescence en présence d’un substrat approprié. L’inhibition prolongée des NO synthases chez ces souris transgéniques engendre une glomérulosclérose qui est prévenue par le losartan, un antagoniste des récepteurs AT1 de l’angiotensine II (ARAII), à une dose non hypotensive. Cet effet protecteur de l’ARAII est associé à un blocage précoce et durable de la stimulation du gène du collagène I [5]. L’activation de ce gène requiert vraisemblablement l’intervention de deux facteurs paracrines synthétisés sous l’influence de l’Ang II, l’endothéline et le TGFβ, puisque nous avons démontré que le bosentan, un antagoniste mixte des récepteurs de l’endothéline et la décorine, un chélateur du TGFβ inhibaient la stimulation de ce gène en présence d’Ang II [6, 7]. Nous avons, ensuite, identifié les voies de signalisation intracellulaire qui participaient à l’activation du promoteur du gène du collagène I par l’Ang II [8]. Cette action de l’Ang II passe par l’activation de la voie des MAP-kinases et du complexe de transcription activating protein AP-1. Nous nous sommes également intéressés à l’étude du récepteur de l’EGF parce que les effets trophiques des agents vasoconstricteurs sont en partie expliqués par la transactivation des récepteurs des facteurs de croissance. Nous avons observé que la phosphorylation du récepteur de l’EGF était nécessaire à la réponse contractile et à l’activation immédiate du gène du collagène de type I par l’endothéline dans des aortes fraîchement isolées de souris transgéniques col Iα2 [9]. À la suite de ces résultats, nous avons constaté que le développement de la fibrose rénale était associé à l’activation du récepteur de l’EGF et de la voie des MAP-kinases dans le cortex rénal. L’inhibition de l’activation du récepteur de l’EGF par le gefitinib, un inhibiteur spécifique de ce récepteur, prévient le développement de l’insuffisance rénale chez l’animal [10]. L’ensemble de nos données sur les mécanismes d’activation du gène du collagène I par l’Ang II est représenté sur la Figure 1.
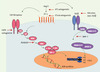 |
Figure 1. Voies de signalisation par lesquelles l’Ang II peut stimuler le gène du collagène I et induire une fibrose rénale vasculaire. |
Régression de la fibrose rénale d’origine vasculaire
Ces dernières années, quelques études expérimentales, mais également cliniques, ont témoigné de la régression des lésions histologiques de néphropathies chroniques fibrosantes. L’observation commune à l’ensemble de ces études est la diminution de la matrice excédentaire glomérulaire et/ou interstitielle [11–15].
Récemment, nous avons constaté la réversibilité des lésions de glomérulosclérose induites par l’inhibition des NO synthases par le L-NAME, chez des souris col Iα2 traitées par un antagoniste mixte des récepteurs de l’endothéline [12]. Parce que la dégradation de l’excédent de matrice déposée requiert l’intervention d’un mécanisme catalytique, nous avons étudié dans ce modèle la cinétique des modifications des collagènes I et IV et de l’activité des enzymes qui assurent leur catabolisme, les MMP 2 et 9, avant et après un traitement antihypertenseur par un ARA II [13]. Les MMP 2 et 9 ont comme substrat préférentiel le collagène de type IV, constituant principal des membranes basales du rein. Après 4 semaines d’hypertension artérielle, les rats présentent une insuffisance rénale, une protéinurie abondante, une expression accrue des collagènes I et IV ainsi que du TFGβ, associées à des lésions de glomérulosclérose. Dans le surnageant des glomérules et des artérioles préglomérulaires isolés à ce stade de la néphropathie, l’activité des MMP 2 et 9 est augmentée. En dépit d’un contrôle incomplet de l’élévation de la pression artérielle systémique, l’ajout de losartan au L-NAME, pendant 4 semaines supplémentaires, normalise la fonction rénale, l’expression des collagènes et du TFGβ et corrige les lésions de glomérulosclérose. À partir de l’étude détaillée de la phase de régression des lésions, nous avons cherché à élucider les mécanismes contribuant au remodelage de la matrice. Celui-ci s’explique par un arrêt précoce de la synthèse des collagènes I et IV sous l’action de l’ARAII tandis que la dégradation de la matrice extracellulaire en excès est suggérée par la stimulation de l’activité in situ des gélatinases (MMP 2 et 9). L’évolution parallèle de la quantité de matrice présente et de l’activité gélatinase plaide en faveur d’une activation de ces enzymes par leurs substrats.
Dans notre étude, nous avons donc observé une disparition complète des lésions histologiques de glomérulosclérose et, conjointement, une normalisation de la fonction rénale et de la protéinurie. Ces résultats s’expliquent vraisemblablement par la brève durée de progression de la glomérulosclérose (pendant les 8 semaines d’administration de L-NAME) et son caractère modéré avec un nombre limité de glomérules détruits, dits « en pain à cacheter ». Si les glomérules peuvent en partie régénérer grâce à la prolifération des cellules endothéliales et mésangiales, les podocytes constituent le facteur limitant du retour à la normale de cette structure. Cette notion a été bien documentée dans le modèle de réduction néphronique (néphrectomie aux 5/6es) chez le rat. De très fortes doses d’énalapril, un inhibiteur de l’enzyme de conversion, administrées à distance de la chirurgie réductrice, entraînent une amélioration des lésions de glomérulosclérose, des lésions tubulo-interstitielles et vasculaires. Mais ni le nombre, ni la taille des podocytes ne sont affectés par ce traitement [14]. Ces constatations soulignent l’importance du dépistage et de la prise en charge précoce des néphropathies chroniques avant l’atteinte d’un point de non-retour.
Conclusions
Les données récentes concernant les mécanismes impliqués dans les altérations structurales de l’insuffisance rénale chronique nous ont permis de faire les constatations suivantes : l’accumulation de la matrice extracellulaire dans le parenchyme rénal est un phénomène réversible ; le degré de réversibilité dépend du degré d’évolution de la néphropathie et de progression de la fibrose. Des traitements pharmacologiques efficaces ont été mis en évidence chez l’animal qui reposent essentiellement sur l’inhibition de l’action de l’Ang II et des systèmes que ce peptide stimule (Figure 1). Il nous semble donc approprié de tester l’efficacité d’un bloqueur de l’Ang II chez les patients de maladie de Fabry. Dans ce sens, des résultats préliminaires encourageants ont été récemment rapportés : la substitution enzymatique couplée avec l’inhibition de l’enzyme de conversion de l’Ang II a stabilisé la fonction rénale et diminué la protéinurie dans une étude réalisée chez un patient [16].
Références
- Maschio G, Alberti D, Janin G, et al. Effect of the angiotensin-converting-enzyme inhibitor benazepril on the progression of chronic renal insufficiency. The angiotensin-converting-enzyme inhibition in progressive renal insufficiency study group. N Engl J Med 1996; 334 : 939–45. [Google Scholar]
- GISEN. Randomised placebo-controlled trial of effect of ramipril on decline in glomerular filtration rate and risk of terminal renal failure in proteinuric, non-diabetic nephropathy. The GISEN group (gruppo italiano di studi epidemiologici in nefrologia). Lancet 1997; 349 : 1857–63. [Google Scholar]
- Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The collaborative study group. N Engl J Med 1993; 329 : 1456–62. [Google Scholar]
- Nakao N, Yoshimura A, Morita H, et al. Combination treatment of angiotensin-II receptor blocker and angiotensin-converting-enzyme inhibitor in non-diabetic renal disease (Cooperate) : a randomised controlled trial. Lancet 2003; 361 : 117–24. [Google Scholar]
- Boffa JJ, Tharaux PL, Placier S, et al. Angiotensin II activates collagen type I gene in the renal vasculature of transgenic mice during inhibition of nitric oxide synthesis : evidence for an endothelin-mediated mechanism. Circulation 1999; 100 : 1901–8. [Google Scholar]
- Chatziantoniou C, Boffa JJ, Ardaillou R, Dussaule JC. Nitric oxide inhibition induces early activation of type I collagen gene in renal resistance vessels and glomeruli in transgenic mice : role of endothelin. J Clin Invest 1998; 101 : 2780–9. [Google Scholar]
- Fakhouri F, Placier S, Ardaillou R, et al. Angiotensin II activates collagen type I gene in the renal cortex and aorta of transgenic mice through interaction with endothelin and TGF-beta. J Am Soc Nephrol 2001; 12 : 2701–10. [Google Scholar]
- Tharaux PL, Chatziantoniou C, Fakhouri F, et al. Angiotensin II activates collagen I gene through a mechanism involving the MAP/ER kinase pathway. Hypertension 2000; 36 : 330–6. [Google Scholar]
- Flamant M, Tharaux PL, Placier S et al. Epidermal growth factor transactivation mediates the tonic and fibrogenic effects of endothelin in the aortic wall of transgenic mice. FASEB J 2003; 17 : 327–9. [Google Scholar]
- François H, Placier S, Parvez-Braun L, et al. Prevention of renal vascular and glomerular fibrosis by inhibiting tyrosine-kinase growth factor receptors. FASEB J 2004; 18 : 926–8. [Google Scholar]
- Fioretto P, Steffes MW, Sutherland DE, et al Reversal of lesions of diabetic nephropathy after pancreas transplantation. N Engl J Med 1998; 339 : 69–75. [Google Scholar]
- Boffa JJ, Tharaux PL, Dussaule JC, et al. Regression of renal vascular fibrosis by endothelin receptor antagonism. Hypertension 2001; 37 : 490–6. [Google Scholar]
- Boffa JJ, Lu Y, Placier S, et al Regression of renal vascular and glomerular fibrosis: role of angiotensin II receptor antagonism and matrix metalloproteinases. J Am Soc Nephrol 2003; 14 : 1132–44. [Google Scholar]
- Adamczak M, Gross ML, Krtil J, et al. Reversal of glomerulosclerosis after high-dose enalapril treatment in subtotally nephrectomized rats. J Am Soc Nephrol 2003; 14 : 2833–42. [Google Scholar]
- Hotta O, Furuta T, Chiba S, et al. Regression of IgA nephropathy : a repeat biopsy study. Am J Kidney Dis 2002; 39 : 493–502. [Google Scholar]
- Warnock DG. Fabry disease : diagnosis and management, with emphasis on the renal manifestations. Curr Opin Nephrol Hypertens 2005; 14 : 87–95. [Google Scholar]
Liste des figures
|
Figure 1. Voies de signalisation par lesquelles l’Ang II peut stimuler le gène du collagène I et induire une fibrose rénale vasculaire. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.