")
")
| Issue |
Med Sci (Paris)
Volume 21, Number 4, Avril 2005
|
|
|---|---|---|
| Page(s) | 355 - 357 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/2005214355 | |
| Published online | 15 avril 2005 | |
Activer un signal bêta-caténine dans le foie est oncogénique
Activating a beta-catenin signal in the liver is oncogenic
Inserm U.567, CNRS UMR 8104, Institut Cochin, Université Paris V, 24, rue du Faubourg-Saint-Jacques, 75014 Paris, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Associée aux facteurs de risque majeurs que sont les hépatites virales B et C (en augmentation constante dans le monde occidental), et les hépatites alcooliques, une recrudescence de cancers primitifs du foie est actuellement observée. Ces carcinomes hépatocellulaires (CHC) se développent tardivement sur des foies cirrhotiques et sont de mauvais pronostic. Cette carcinogenèse est encore mal comprise à l’échelle moléculaire, et jusqu’il y a quelques années, deux grandes voies de signalisation, p53 et Rb, étaient décrites comme étant fréquemment inactivées. La modélisation chez la souris était prometteuse, permettant d’évaluer, à long terme, l’effet d’une mutation génétique unique. Cependant, la létalité précoce des modèles murins d’inactivation des gènes suppresseurs de tumeurs p53 et Rb ne permit pas d’évaluer le rôle oncogénique de ces invalidations dans le foie [1]. En revanche, des tumeurs hépatiques purent être détectées chez d’autres souris exprimant dans le foie l’oncogène c-myc [2, 3], fréquemment amplifié dans les CHC.
Les mutations béta-caténine définissent une classe particulière de CHC chez l’homme
La voie de signalisation Wnt/béta-caténine joue un rôle clé pour déterminer des choix de destin cellulaire au cours du développement embryonnaire et pour l’autorenouvellement de cellules souches chez l’adulte : dans ces deux cas, les facteurs sécrétés Wnt sont capables d’induire une cascade d’événements dont la béta-caténine est l’effecteur principal (Figure 1). En 1997, cette signalisation fut impliquée dans la cancérogenèse puisqu’il était découvert que le gène Apc (adenomatous polyposis coli) devait son rôle suppresseur de tumeur à sa participation à la dégradation de la béta-caténine [4, 5]. Apc étant inactivé dans plus de 80 % des cancers colorectaux, une activation aberrante d’un signal béta-caténine était donc l’événement majeur mais aussi initiateur de ces cancers. Tirant parti de ces résultats, il fut montré dans l’équipe que des mutations du gène codant pour la béta-caténine, stabilisatrices de la protéine et l’entraînant donc vers une signalisation aberrante et continue, était rencontrée dans 25 % des CHC chez l’homme, et 50 % des CHC chez les souris sur-exprimant c-myc [6]. Il est maintenant admis qu’une activation du signal béta-caténine, mise en évidence par son accumulation cytosolique et/ou nucléaire (Figure 2) concerne 30 à 40 % des CHC chez l’homme [7] : elle est due principalement à des mutations du gène béta-caténine lui-même, mais peuvent survenir également des inactivations plus rares du gène suppresseur de tumeur AXIN1, alors que les cas d’inactivation d’APC sont exceptionnels.
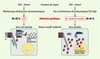 |
Figure 1. Voie béta-caténine et son activation aberrante dans les carcinomes hépatocellulaires. Les facteurs de risque que sont les hépatites virales B (HVB) et les hépatites virales C (HVC) ou la prise d’alcool, définissent deux voies de carcinogenèse hépatique. Aux altérations génétiques inactivant la voie p53 (perte 17p, mutations p53) correspond globalement une voie béta-caténine non activée, où la béta-caténine ne joue un rôle que dans l’adhérence cellulaire en liant la E-cadhérine (E-cadh) au cytosquelette d’actine : dans ce cas, un complexe multiprotéique, orchestré par l’axine et incluant APC, permet la phosphorylation de la béta-caténine (béta) par la kinase GSK3béta, puis son ubiquitinylation et sa dégradation par le protéasome. Dans un contexte où la béta-caténine est mutée ou délétée (béta*) au niveau de ses sites de phosphorylation par GSK3béta, une activation constitutive du signal béta-caténine se produit : la béta-caténine ne peut plus être dégradée, s’accumule dans le noyau où, associée aux facteurs de transcription de la famille LEF/TCF (TCF), elle induit la transcription de gènes cibles. Les fréquences (%) des mutations géniques p53 et béta-caténine dans les CHC sont indiquées. |
 |
Figure 2. Hépatocarcinogenèse à la suite de l’invalidation hépatospécifique et ménagée d’Apc dans le foie. A. À partir de souris flanquées de sites loxP sur l’exon 14 des deux allèles du gène Apc (Apclox), la délétion de cet exon 14 (Apcdelta14) peut être obtenue par action ciblée d’une recombinase Cre : elle permet la production d’un codon stop prématuré, empêchant l’exon 15, qui code normalement pour les domaines de dégradation de la béta-caténine, d’être traduit. B. L’injection intraveineuse d’adénovirus Cre chez les souris Apclox permet un ciblage préférentiel du foie. À forte dose adénovirale, 90 % des hépatocytes présentent une invalidation bi-allélique d’Apc, qui conduit à l’activation d’un signal béta-caténine, mise en évidence par l’accumulation cytosolique et nucléaire de béta-caténine. Les conséquences sont létales et un phénotype sévère d’hépatomégalie est constaté chez ces souris. À la suite d’une infection par des doses plus faibles d’adénovirus Cre, environ 30 % des hépatocytes présentent une invalidation du gène, mise en évidence par une immunocytochimie de la béta-caténine. Cette invalidation, compatible avec la survie des animaux, permet le développement d’hépatocarcinomes en 8 à 9 mois. |
Le groupe de J. Zucman-Rossi a pu montrer que la voie béta-caténine, quand elle est activée par une mutation du gène béta-caténine, définissait une voie de cancérogenèse particulière survenant en dehors d’un contexte d’hépatite virale B et surtout associée à peu d’instabilités chromosomiques [8] (Figure 1). Les deux autres grandes voies, notamment celle de p53, intervenant dans le cadre d’une hépatite virale B, sont, quant à elles, associées à un contexte chomosomique très instable faisant intervenir de nombreuses pertes alléliques, favorisant la perte de gènes suppresseurs de tumeurs.
Le signal béta-caténine est-il en cause dans la survenue de CHC ?
C’est bien entendu ce qu’il restait à démontrer, et c’est ce à quoi s’employa notre groupe ces dernières années, tout d’abord en développant un modèle murin de transgenèse additionnelle où l’expression d’un mutant stable de béta-caténine était ciblée dans le foie. Malheureusement, ces souris mouraient rapidement à la suite du développement d’une hépatomégalie importante [9].
Une collaboration avec M. Giovannini nous permit alors de créer un modèle d’invalidation hépato-spécifique bi-allélique du gène Apc, en utilisant la stratégie Cre-loxP (Figure 2) [10]. Par l’injection intraveineuse d’une forte dose d’adénovirus codant pour la recombinase Cre, nous étions capables d’invalider Apc et d’activer la signalisation béta-caténine dans quasiment tous les hépatocytes. Cette invalidation massive reproduisait strictement le phénotype d’hépatomégalie et de mort rapide des souris exprimant un mutant oncogénique béta-caténine. Ainsi, Apc était bien fonctionnel dans le tissu hépatique, posant la question, à ce jour encore sans réponse, de l’absence de mutations de ce gène dans les CHC chez l’homme. Avec une dose moitié moindre d’adénovirus Cre, un signal béta-caténine put être détecté dans 30 % à 40 % des hépatocytes, ce qui était compatible avec la survie des animaux. Une réaction immunitaire due à l’infection adénovirale induisait néanmoins la disparition de nombreux hépatocytes Apc−/−, mais il en subsistait de 1 à 6 ‰ à long terme dans le foie, qui étaient capables de donner naissance en neuf mois à des CHC présentant, comme chez l’homme, divers degrés de différenciation. Ce modèle murin est donc particulièrement pertinent pour l’étude du CHC humain. En effet, contrairement aux modèles transgéniques où l’expression d’oncogènes survient dans tous les hépatocytes, les hépatocytes Apc−/−, par leur dispersion au sein d’un foie génétiquement non altéré, reproduisent un phénomène clonal de mutation, classiquement initiateur d’une carcinogenèse. Mais, ce modèle montre aussi qu’un événement fréquent lors de la cancérogenèse hépatique humaine, c’est-à-dire l’activation constitutive du signal béta-caténine, est bien capable de conduire au développement de carcinomes hépatocellulaires chez la souris.
Quels sont les conséquences moléculaires du signal béta-caténine et quels événements génétiques coopèrent avec la béta-caténine pour induire la formation de CHC ?
Il reste maintenant un vaste terrain d’investigations qui visent à établir une chronologie dans les différents événements génétiques participant à cette carcinogenèse. Il s’agira donc sur le modèle murin Apc−/− d’évaluer le transcriptome et l’intégrité du génome hépatique, à court terme après activation du signal béta-caténine, dans le temps de latence de 6 mois pendant lequel l’hépatocyte résiduel Apc−/− reste quiescent, dans les micronodules et dans les CHC établis. Cela devrait permettre de comprendre le processus multi-étapes de cette carcinogenèse hépatique au cours de laquelle la voie de la béta-caténine est activée, et d’ouvrir peut-être le champ vers de nouvelles cibles thérapeutiques.
Références
- Hooper ML. The role of the p53 and Rb-1 genes in cancer, development and apoptosis. J Cell Sci 1994; 18 (suppl) : 13–7. [Google Scholar]
- Cartier N, Miquerol L, Tulliez M, et al. Diet-dependent carcinogenesis of pancreatic islets and liver in transgenic mice expressing oncogenes under the control of the L-type pyruvate kinase gene promoter. Oncogene 1992; 7 : 1413–22. [Google Scholar]
- Etiemble J, Degott C, Renard CA, et al. Liver-specific expression and high oncogenic efficiency of a c-myc transgene activated by woodchuck hepatitis virus insertion. Oncogene 1994; 9 : 727–37. [Google Scholar]
- Korinek V, Barker N, Morin PJ, et al. Constitutive transcriptional activation by a béta-catenin-Tcf complex in APC−/− colon carcinoma. Science 1997; 275 : 1784–7. [Google Scholar]
- Rubinfeld B, Albert I, Porfiri E, et al. Binding of GSK3béta to the APC-béta-catenin complex and regulation of complex assembly. Science 1996; 272 : 1023–6. [Google Scholar]
- De La Coste A, Romagnolo B, Billuart P, et al. Somatic mutations of the béta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc Natl Acad Sci USA 1998; 95 : 8847–51. [Google Scholar]
- Buendia MA. Genetics of hepatocellular carcinoma. Semin Cancer Biol 2000; 10 : 185–200. [Google Scholar]
- Laurent-Puig P, Legoix P, Bluteau O, et al. Genetic alterations associated with hepatocellular carcinomas define distinct pathways of hepatocarcinogenesis. Gastroenterology 2001; 120 : 1763–73. [Google Scholar]
- Cadoret A, Ovejero C, Saadi-Kheddouci S, et al. Hepatomegaly in transgenic mice expressing an oncogenic form of béta-catenin. Cancer Res 2001; 61 : 3245–9. [Google Scholar]
- Colnot S, Decaens T, Niwa-Kawakita M, et al. Liver-targeted disruption of Apc in mice activates béta-catenin signaling and leads to hepatocellular carcinomas. Proc Natl Acad Sci USA 2004; 101 : 17216–21. [Google Scholar]
© 2005 médecine/sciences - Inserm / SRMS
Liste des figures
|
Figure 1. Voie béta-caténine et son activation aberrante dans les carcinomes hépatocellulaires. Les facteurs de risque que sont les hépatites virales B (HVB) et les hépatites virales C (HVC) ou la prise d’alcool, définissent deux voies de carcinogenèse hépatique. Aux altérations génétiques inactivant la voie p53 (perte 17p, mutations p53) correspond globalement une voie béta-caténine non activée, où la béta-caténine ne joue un rôle que dans l’adhérence cellulaire en liant la E-cadhérine (E-cadh) au cytosquelette d’actine : dans ce cas, un complexe multiprotéique, orchestré par l’axine et incluant APC, permet la phosphorylation de la béta-caténine (béta) par la kinase GSK3béta, puis son ubiquitinylation et sa dégradation par le protéasome. Dans un contexte où la béta-caténine est mutée ou délétée (béta*) au niveau de ses sites de phosphorylation par GSK3béta, une activation constitutive du signal béta-caténine se produit : la béta-caténine ne peut plus être dégradée, s’accumule dans le noyau où, associée aux facteurs de transcription de la famille LEF/TCF (TCF), elle induit la transcription de gènes cibles. Les fréquences (%) des mutations géniques p53 et béta-caténine dans les CHC sont indiquées. |
| Dans le texte | |
|
Figure 2. Hépatocarcinogenèse à la suite de l’invalidation hépatospécifique et ménagée d’Apc dans le foie. A. À partir de souris flanquées de sites loxP sur l’exon 14 des deux allèles du gène Apc (Apclox), la délétion de cet exon 14 (Apcdelta14) peut être obtenue par action ciblée d’une recombinase Cre : elle permet la production d’un codon stop prématuré, empêchant l’exon 15, qui code normalement pour les domaines de dégradation de la béta-caténine, d’être traduit. B. L’injection intraveineuse d’adénovirus Cre chez les souris Apclox permet un ciblage préférentiel du foie. À forte dose adénovirale, 90 % des hépatocytes présentent une invalidation bi-allélique d’Apc, qui conduit à l’activation d’un signal béta-caténine, mise en évidence par l’accumulation cytosolique et nucléaire de béta-caténine. Les conséquences sont létales et un phénotype sévère d’hépatomégalie est constaté chez ces souris. À la suite d’une infection par des doses plus faibles d’adénovirus Cre, environ 30 % des hépatocytes présentent une invalidation du gène, mise en évidence par une immunocytochimie de la béta-caténine. Cette invalidation, compatible avec la survie des animaux, permet le développement d’hépatocarcinomes en 8 à 9 mois. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.