")
")
| Issue |
Med Sci (Paris)
Volume 20, Number 12, Décembre 2004
|
|
|---|---|---|
| Page(s) | 1066 - 1068 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/200420121066 | |
| Published online | 15 décembre 2004 | |
L’amaurose congénitale de Leber : les rétinol-déshydrogénases au banc des accusés
Leber congenital amaurosis: retinol dehydrogenases are incriminated
Inserm U.393, Handicaps génétiques de l’enfant, Hôpital Necker-Enfants Malades, 149, rue de Sèvres, 75743 Paris Cedex 15, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Il est désormais admis que l’amaurose congénitale de Leber (ACL) [MIM 204000] [1, 2] est la dystrophie rétinienne la plus sévère et la plus précoce puisqu’elle est responsable de cécité ou de malvoyance profonde néonatale. Nous avons démontré récemment que, sous ce vocable, se distinguaient deux groupes de maladies déterminées génétiquement, se transmettant à une exception près selon le mode récessif autosomique [3]. Le premier groupe correspond à la description faite par A. Sorsby et C.E. Williams qui n’hésitaient pas à parler d’« aplasie rétinienne » [4]. Nous avons effectivement retrouvé cette entité chez des individus atteints d’une forme très sévère, très précoce et non évolutive dans laquelle l’atteinte des cônes est prédominante. Cette forme est désignée par, selon la terminologie anglo-saxonne, cone-rod dystrophy. Le second groupe correspond à l’extrémité d’un spectre de gravité des rétinopathies pigmentaires et est constitué d’individus souffrant d’une dystrophie, certes sévère, mais plus progressive, débutant par une atteinte des bâtonnets (rod) et désignée selon la même terminologie par rod-cone dystrophy [5]. Pour les deux groupes, les critères cliniques d’inclusion du diagnostic sont un nystagmus congénital à grandes oscillations, une absence de poursuite oculaire, des signes digito-oculaires ainsi qu’un fond d’œil normal à la naissance qui contraste avec un ERG (électrorétinogramme) plat, témoignant d’une atteinte des deux types de photorécepteurs.
La distinction entre les deux groupes cliniques se fait à la fin de la première année de vie et repose essentiellement sur : (1) l’étude du comportement de l’enfant à la lumière ; (2) l’aspect de la rétine ; (3) les données précoces de la réfraction ; et (4) l’acuité visuelle (Figure 1).
 |
Figure 1. Représentation graphique des corrélations génotype-phénotype établies pour l’amaurose congénitale de Leber et fréquence de l’implication de chaque gène responsable de la maladie. |
Jusqu’à une période très récente, 10 gènes codant pour l’ACL étaient localisés et 7 identifiés : GUCY2D [MIM 600179], RPE65 [MIM 180069], CRX [MIM 600225], AIPL1 [MIM 604392], RPGRIP1 [MIM 605446], CRB1 [MIM 604210], TULP1 [602280], LCA3 [MIM 604232] en 14q24, LCA5 [MIM 604537] en 6q11-16 et LCA9 en 1p36 [MIM 608553]. Ces gènes sont tous exprimés préférentiellement dans les photorécepteurs ou l’épithélium pigmentaire de la rétine, mais ils sont impliqués dans des mécanismes physiologiques extraordinairement différents, entraînant une variabilité physiopathogénique inattendue.
Afin d’identifier de nouveaux gènes responsables de la maladie, nous avons choisi de tester plusieurs gènes connus codant pour des protéines de la cascade des rétinoïdes, qui permet aux pigments visuels de se régénérer après stimulation lumineuse. En effet, deux gènes de cette cascade, RPE65 et LRAT, ont été décrits comme responsables d’ACL ou d’une dystrophie rétinienne sévère de l’enfant (CSRD) [6–8]. Les déshydrogénases rétiniennes (RDH), qui ont un rôle crucial dans la conversion de la vitamine A au cours du cycle visuel - RDH10 et RDH11 spécifiquement exprimés au niveau de l’épithélium pigmentaire, RDH8, RDH12-14 spécifiques des photorécepteurs - nous ont paru être de bons candidats [9]. Tous ces gènes ont donc été étudiés dans une cohorte de 110 patients atteints d’ACL pour lesquels les 7 gènes avaient été exclus par analyse directe ainsi que les trois locus pour les formes familiales. Aucun des 110 patients ne porte de mutations dans les gènes RDH 8, 10, 11, 13 et 14.
En revanche, 11 mutations du gène RDH12 ont été identifiées chez 8 parmi les 110 patients, mutations dont la ségrégation familiale a été vérifiée pour chacune des 8 familles (Figure 2). Il faut souligner que toutes ces mutations ont été identifiées chez des patients atteints de la forme rod-cone dystrophy. Ce groupe est constitué de 44/110 individus testés. Ainsi, les mutations du gène RDH12 rendent compte de 18 % de ces cas [1].
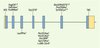 |
Figure 2. Structure du gène RDH12 et mutations identifiées comme responsables d’amaurose congénitale de Leber et de dystrophie rétinienne sévère de l’enfant. |
Parallèlement à notre étude, l’équipe d’Andreas Gal (Hambourg, Allemagne) a publié une étude relatant l’implication du gène RDH12 dans la CSRD [2]. Leur étude a débuté par l’identification d’une liaison génétique au locus 14q23.3-q24 pour trois familles originaires d’Autriche atteintes d’une CSRD. Le gène RDH12 contenu dans cet intervalle génétique s’est avéré être un bon candidat par sa fonction dans le cycle visuel. Cette étude a permis d’identifier une mutation ancestrale responsable de CSRD dans les trois familles autrichiennes (Tyr226Cys, Figure 2). Une étude extensive dans des familles atteintes de dystrophies sévères de l’enfant (CSRD ou ACL) a permis d’identifier 4 autres mutations dans trois familles non autrichiennes. Une étude fonctionnelle a permis de valider les mutations faux-sens décrites comme causales puisqu’elles sont responsables d’une baisse de l’activité enzymatique permettant la conversion du tout-trans rétinol en tout-trans rétinal. RDH12, bien qu’appartenant à une famille de gènes, semble avoir un rôle unique dans les cellules photoréceptrices. L’étude de liaison génétique effectuée par A.R. Janecke et al. dans les trois familles autrichiennes avait suggéré que RDH12 était peut-être le gène correspondant à LCA3 [2]. Néanmoins, David Stockton nous a confirmé qu’aucune mutation de RDH12 n’avait été retrouvée dans la famille princeps [10].
En résumé, les travaux menés par A.R. Janecke et al. - et par notre laboratoire - permettent de dégager trois conclusions : la première est que, une fois encore, des patients atteints d’une ACL non ambiguë sont porteurs de mutations dans un gène impliqué également dans des rétinopathies pigmentaires précoces de l’enfant. La seconde est que la place du gène RDH12 au sein des gènes responsables de la maladie n’est pas anecdotique puisqu’il rend compte de 4,5 % du total des patients. La troisième est que, en dépit de leur fonction enzymatique commune, les autres RDH ne complémentent pas le déficit en RDH12 et qu’aucune autre ne semble impliquée dans l’ACL. Enfin, cette étude tend à démontrer que la fréquence de la maladie, toutes formes confondues, estimée autrefois à 5 % de l’ensemble des dystrophies rétiniennes héréditaires, est certainement très sous-estimée.
Références
- Perrault I, Hanein S, Gerber S, et al. Retinal dehydrogenase 12 (RDH12) mutations in Leber congenital amaurosis. Am J Hum Genet 2004; 75 : 639–46. [Google Scholar]
- Janecke AR, Thompson DA, Utermann G, et al. Mutations in RDH12 encoding a photoreceptor cell retinol dehydrogenase cause childhood-onset severe retinal dystrophy. Nat Genet 2004; 36 : 850–4. [Google Scholar]
- Perrault I, Hanein S, Gerber S, et al. Evidence of autosomal dominant Leber congenital amaurosis (LCA) underlain by a CRX heterozygous null allele. J Med Genet 2003; 40 : e90. [Google Scholar]
- Sorsby A, Williams CE. Retinal aplasia as a clinical entity. Br Med J 1960; 1 : 293–97. [Google Scholar]
- Hanein S, Perrault I, Gerber S, et al. Leber congenital amaurosis : comprehensive survey of the genetic heterogeneity, refinement of the clinical definition, and genotype-phenotype correlations as a strategy for molecular diagnosis. Hum Mutat 2004; 23 : 306–17. [Google Scholar]
- Marlhens F, Bareil C, Griffoin JM, et al. Mutations in RPE65 cause Leber’s congenital amaurosis. Nat Genet 1997; 17 : 139–41. [Google Scholar]
- Gu SM, Thompson DA, Srikumari CR, et al. Mutations in RPE65 cause autosomal recessive childhood-onset severe retinal dystrophy. Nat Genet 1997; 17 : 194–7. [Google Scholar]
- Thompson DA, Li Y, McHenry CL, et al. Mutations in the gene encoding lecithin retinol acyltransferase are associated with early-onset severe retinal dystrophy. Nat Genet 2001; 28 : 123–4. [Google Scholar]
- Kuska V, Imanishi Y, Batten M, et al. Retinoid cycle in the vertebrate retina : experimental approaches and mechanisms of isomerization. Vision Res 2003; 43 : 2959–81. [Google Scholar]
- Stockton DW, Lewis RA, Abboud EB, et al. A novel locus for Leber congenital amaurosis on chromosome 14q24. Hum Genet 1998; 103 : 328–33. [Google Scholar]
© 2004 médecine/sciences - Inserm / SRMS
Liste des figures
|
Figure 1. Représentation graphique des corrélations génotype-phénotype établies pour l’amaurose congénitale de Leber et fréquence de l’implication de chaque gène responsable de la maladie. |
| Dans le texte | |
|
Figure 2. Structure du gène RDH12 et mutations identifiées comme responsables d’amaurose congénitale de Leber et de dystrophie rétinienne sévère de l’enfant. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.