")
")
| Issue |
Med Sci (Paris)
Volume 20, Number 11, Novembre 2004
|
|
|---|---|---|
| Page(s) | 954 - 956 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/20042011954 | |
| Published online | 15 novembre 2004 | |
Le syndrome de Cornelia de Lange
Le syndrome de Cornelia de Lange
9, rue Basse, 54330 Clerey-sur-Brenon, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Le syndrome de Cornelia de Lange (CdLS) est un peu la lanterne rouge des retards mentaux syndromiques, puisque le gène impliqué dans cette maladie génétique vient seulement d’être découvert. Ce syndrome est pourtant connu depuis des décennies et sa symptomatologie est caractéristique, mais la ressemblance clinique avec le syndrome dup (3q) a fait quelque peu errer la découverte du locus que l’on supposait sur le chromosome 3.
Un peu d’histoire
C’est en 1933 que Cornelia de Lange, professeur de pédiatrie à Amsterdam individualisa le syndrome qui porte son nom… et son prénom, car il était exceptionnel à l’époque qu’une femme identifiât un nouveau syndrome. En fait, Cornelia n’était pas la première à avoir décrit cette maladie congénitale qu’elle avait appelé le typus amstelodamensis. Personne, sans doute, ne s’en serait rendu compte si John Marius Opitz, généticien d’origine allemande, fort érudit et grand pourvoyeur d’éponymes, ne l’avait redécouvert trente ans plus tard tout à fait par hasard : à l’université d’Utah, la rupture d’une canalisation d’eau avait inondé la bibliothèque. La responsable, pour lui faire part du désastre, lui mit sous les yeux de vieux tomes très abimés du Jahrbuch für Kinderheilkunde und physische Erziehung. Quelle ne fut pas la surprise d’Opitz en retrouvant, juste avant les feuilles qui avaient été agglutinées, une excellente description du CdLS faite en 1916 par un jeune médecin allemand, Winfried R.C. Brachmann. Ce dernier s’excusait, à la fin de son texte, de ne pouvoir poursuivre ses travaux. Mobilisé dans l’armée allemande, son temps était venu de partir à la guerre. Il semble qu’il n’en soit pas revenu. Opitz insista donc pour ajouter au CdLS ce nouvel éponyme, Brachmann, sans y réussir totalement [1].
Données cliniques et épidémiologiques
Le phénotype du CdLS est cliniquement évident en raison de l’aspect caractéristique du visage : synophris1, implantation basse des cheveux sur le front et dans la nuque avec hirsutisme1, ensellure nasale profonde, dents petites et espacées, oreilles bas implantées. S’y associent des anomalies des membres pouvant aller d’une discrète malformation des pouces à une phocomélie1. Les anomalies radiologiques, retard de maturation osseuse, microbrachycéphalie, anomalies thoraciques et réduction des angles acétabulaires1 complètent le tableau clinique. Malgré la grande variabilité des signes, le diagnostic de ce retard mental syndromique, transmis en dominance mais observé le plus souvent à l’état isolé dans les familles, est relativement facile, d’autant qu’il n’est pas exceptionnel (1/50 000 naissances environ).
Un gène qui vient de loin
Pourtant, la recherche du locus fut laborieuse, en raison de plusieurs cas de CdLS avec remaniements chromosomiques, en particulier sur le bras long du chromosome 3. Ce n’est donc que tout récemment que le locus a été trouvé, en 5p13, et le gène identifié [2, 3]. Appelé NIPBL (nipped-B like), il code pour une protéine qui a été baptisée delangine. Chez la drosophile, le gène Nipped-B est un régulateur majeur de la voie Notch et des gènes Cut et Ultrabithorax. La protéine codée par Nipped-B appartient à la famille des adhérines chromosomiques qui, en modifiant l’architecture, facilite à longue distance les interactions entre les promoteurs et les séquences stimulatrices. Ubx réprime le gène Dll (distalless) indispensable au développement des membres. Il est donc possible que les troubles du développement des membres dans le CdLS soient en relation avec une anomalie de régulation des gènes DLX (homologues de Dll de la drosophile chez les mammifères). Grâce à une recherche in silico, des homologues de NIPBL ont été retrouvés dans de nombreuses espèces, en particulier dans les levures où ils appartiennent à la famille des Scc2 intervenant dans la cohésion des chromatides sœurs. Une étude récente vient du reste de démontrer le double rôle de Nipped-B chez la drosophile : il intervient à la fois en maintenant la cohésion des chromatides sœurs et en facilitant l’activation à distance du gène Cut [4].
Le type de mutations observées chez les sujets atteints de CdLS semble avoir pour conséquence la production d’une protéine tronquée, ou la perte de celle-ci. La maladie est donc due à une haplo-insuffisance1. L’observation d’un enfant porteur d’une grande délétion de la région, englobant le gène NIPBL, et ayant des manifestations cliniques particulièrement sévères corrobore cette hypothèse. Sur la cinquantaine de cas étudiés, 50 % environ sont porteurs d’une mutation. Il est encore trop tôt pour dire s’il existe une hétérogénéité génétique ou si certaines mutations dans le gène NIPBL n’ont pu être mises en évidence.
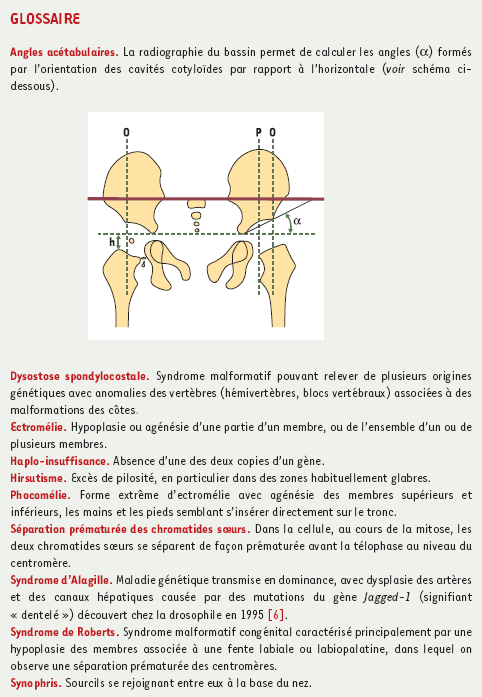
Comme il existe une ressemblance clinique entre le syndrome de Roberts1 (OMIM 268300) et le CdLS, les auteurs [2] ont eu l’idée de rechercher dans les cellules des patients atteints de CdLS une séparation prématurée des chromatides sœurs1 (SCP) : l’examen des mitoses en bandes C s’est révélé négatif. Toutefois, on pouvait peut-être s’y attendre. Le syndrome de Roberts est récessif, et il faudrait donc plutôt étudier un modèle animal avec invalidation du gène à l’état homozygote pour voir si la perte des deux copies de Nipbl entraîne une SCP.
Que le gène NIPBL soit en cause dans le CdLS, maladie multisystémique, apparaît parfaitement logique. D’autres gènes impliqués dans la voie Notch [5] sont responsables de maladies humaines, comme JAG1 dans le syndrome d’Alagille1 (→) ou DLL3 dans la dysostose spondylocostale1 (→). Cette découverte, bien que tardive, devrait permettre de comprendre le rôle de la delangine au cours du développement embryonnaire et d’enrichir les connaissances de la voie de signalisation Notch chez l’homme.
(→) m/s 1997, n° 2, p. 1481
(→) m/s 2000, n° 8-9, p. 1000
Voir Glossaire.
Références
- Opitz JM. The Brachmann-de Lange syndrome. Am J Med Genet 1985; 22 : 89–102. [Google Scholar]
- Tonkin ET, Wang TJ, Lisgo S, et al. NIPBL, encoding a homolog of fungal Scc2-type sister chromatid cohesion proeins and fly Nipped-B is mutated in Cornelia de Lange syndrome. Nat Genet 2004; 36 : 631–5. [Google Scholar]
- Krantz ID, McCallum J, DeScipio C, et al. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nat Genet 2004; 36 : 636–41. [Google Scholar]
- Rollins RA, Korom M, Aulner N, et al. Drososphila Nipped-B protein supports sister chromatid cohesion and opposes the stromalin/Scc3 cohesion factor to facilitate long-range activation of the cut gene Mol Cell Biol 2004; 24 : 3100–11. [Google Scholar]
- Schweisguth F. Fonctions et régulation de l’activité de signalisation du récepteur Notch. Med Sci (Paris) 2000;16 :186–91. [Google Scholar]
- Lindsell CE, Shawber CJ, Boulter J, Weinmaster G. Jagged : a mammalian ligand that activates Notch1. Cell 1995; 80 : 909–17. [Google Scholar]
© 2004 médecine/sciences - Inserm / SRMS
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.