")
")
| Issue |
Med Sci (Paris)
Volume 20, Number 11, Novembre 2004
|
|
|---|---|---|
| Page(s) | 952 - 954 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/20042011952 | |
| Published online | 15 novembre 2004 | |
La huntingtine stimule le transport du BDNF
Stimulation of BDNF transport by huntingtin
UMR 146 CNRS, Institut Curie, Bâtiment 110, Centre Universitaire, 91405 Orsay Cedex, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
L’expansion anormale de glutamines (polyQ) dans la protéine huntingtine est à l’origine de la maladie de Huntington, qui affecte plus de 6 000 personnes en France. Cette maladie est caractérisée par des mouvements incontrôlés (chorée) et des désordres psychiques et intellectuels conduisant à une incapacité totale, à la démence et, finalement, à la mort des patients. La lésion neuropathologique dans la maladie de Huntington implique le dysfonctionnement et la dégénérescence de certains neurones du cerveau, en particulier des neurones du striatum, une structure impliquée dans le contrôle du mouvement. Malgré de nombreuses études, la fonction de la huntingtine normale ainsi que les mécanismes moléculaires par lesquels la huntingtine polyQ (mutante, contenant l’expansion anormale) conduit au dysfonctionnement et à la mort neuronale ne sont pas encore bien compris [1]. Les travaux de L.R. Gauthier et al. [2] apportent de nouveaux éléments pour comprendre ces mécanismes et décrivent pour la première fois une fonction pour la huntingtine dans le transport de vésicules contenant un facteur neurotrophique, le brain-derived neurotrophic factor (BDNF). En situation pathologique, cette fonction est altérée, ce qui conduit à la dégénérescence des neurones du striatum.
Le BDNF, un facteur neurotrophique d’importance dans la maladie de Huntington
Le BDNF est produit par les neurones du cortex et délivré vers le striatum où il est nécessaire à la différenciation et à la survie des neurones de cette région. Pourquoi étudier le BDNF dans la maladie de Huntington ? In vitro, l’ajout de BDNF inhibe la mort des neurones striataux induite par la huntingtine polyQ. In vivo, les concentrations de BDNF sont diminuées dans le cerveau de patients atteints. Cette diminution de BDNF a d’abord été attribuée à un défaut transcriptionnel [3], mais l’étude de L.R. Gauthier et al. [2] montre qu’elle est également due à un dérèglement du transport vésiculaire et, en conséquence, à un défaut de sa libération dans la synapse.
La huntingtine stimule le transport vésiculaire du BDNF le long des microtubules
Bien qu’on puisse l’observer dans les noyaux, la huntingtine est majoritairement cytoplasmique, associée à des structures vésiculaires et aux microtubules (Figure 1). Les auteurs [2] ont formulé l’hypothèse selon laquelle la huntingtine pourrait influencer le transport intracellulaire du BDNF dans les neurones du cortex (neurones qui produisent le BDNF et le transportent vers le striatum). Pour étudier la dynamique du BDNF, une technique de vidéomicroscopie rapide permettant de suivre et d’analyser en trois dimensions la dynamique intracellulaire de protéines fluorescentes (ici, le BDNF fusionné à la green fluorescent protein) a été utilisée. L’effet des huntingtines sauvage ou polyQ sur la dynamique des vésicules BDNF a été déterminé : la huntingtine normale, mais pas la huntingtine polyQ, conduit à une augmentation de la vitesse des vésicules et à une diminution de leur temps de pause. Cet effet est spécifique de la huntingtine, car le transport de BDNF est diminué lorsque l’expression de la huntingtine est réduite par la technique d’interférence par l’ARN. Quels sont les mécanismes moléculaires impliqués ? La huntingtine interagit avec la huntingtin-associated protein 1 (HAP1) qui, elle-même, interagit avec des protéines impliquées dans le transport vésiculaire le long des microtubules, notamment la sous-unité p150Glued de la dynactine (Figure 2). La huntingtine polyQ, via HAP1, désorganise l’association entre les composants de la machinerie motrice et les microtubules, ce qui conduit à une diminution du transport intracellulaire le long des microtubules. Finalement, l’altération du transport de BDNF résulte en une réduction de sa libération et une augmentation de la mort cellulaire.
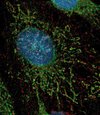 |
Figure 1. La huntingtine et le BDNF sont colocalisés dans des cellules d’origine neuronale. Les cellules sont fixées, immunomarquées pour la huntingtine (en rouge) et pour le BDNF (brain-derived neurotrophic factor) (en vert), puis analysées par microscopie à déconvolution (photo, Jim P. Dompierre, Institut Curie, Orsay, France). |
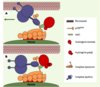 |
Figure 2. Fonction de la huntingtine dans le transport du BDNF. A. La huntingtine normale s’associe au complexe dynéine-dynactine qui est un des moteurs moléculaires responsable du transport de cargos le long des microtubules. Cette association se fait via la protéine HAP1 (huntingtin-associated protein 1) : la huntingtine normale agit alors comme un facteur de processivité pour le transport des vésicules contenant BDNF.B. En situation pathologique, l’association huntingtine/HAP1/dynactine est altérée, ce qui a pour conséquence un détachement des protéines du complexe moteur des microtubules. Dans ces conditions, le transport de BDNF est réduit (schéma réalisé par Fabrice P. Cordelières, Institut Curie, Orsay, France). |
La maladie de Huntington : gain, mais aussi perte de fonction
Le gain d’une fonction toxique de la huntingtine polyQ a été principalement établi dans la maladie de Huntington. Il est lié au fait que cette maladie possède un caractère dominant presque pur : les individus hétérozygotes et homozygotes pour la mutation ont des symptomatologies similaires [4]. L’étude de L.R. Gauthier et al. [2] montre que des mécanismes de perte de fonction de la huntingtine sauvage pourraient également intervenir au cours de la maladie.
Ainsi, une des premières étapes de la maladie serait la perte de la capacité de la huntingtine polyQ de transporter efficacement le facteur neurotrophique BDNF, conduisant alors à une plus grande vulnérabilité des neurones. Une autre étape essentielle est le clivage protéolytique de la huntingtine polyQ, qui conduit à la production de fragments aminoterminaux qui sont transportés dans le noyau et forment des agrégats [5]. Dans le noyau, ces fragments de huntingtine polyQ induisent la mort des neurones par un mécanisme « gain de fonction » qui implique un dérèglement de l’activité transcriptionnelle de facteurs tels que CBP (CREB [cAMP-responsive element-binding protein] binding protein), Sp1 et TAFII130 (TATA-binding protein associated factor) [1, 6].
La protéolyse n’affecte pas seulement la huntingtine polyQ. En effet, la huntingtine sauvage est également clivée chez les patients. De manière intéressante, un fragment aminoterminal (exon 1 de la huntingtine) n’est pas capable de stimuler le transport de BDNF [2]. Ainsi, une des conséquences de la protéolyse de la huntingtine sauvage pourrait être d’inactiver sa fonction dans le transport. Ce clivage chez les patients pourrait, en réduisant le transport de BDNF, augmenter la toxicité induite par l’allèle polyQ.
Une des caractéristiques majeures de la maladie de Huntington est l’agrégation de la huntingtine polyQ dans le noyau comme dans le cytoplasme. Le rôle exact de ces agrégats reste sujet à controverse. Cependant, plusieurs laboratoires ont montré que les agrégats neuritiques de huntingtine polyQ bloquent de façon physique le transport axonal [7]. Ainsi, dans les stades précoces de la maladie, la fonction de la huntingtine dans le transport serait perdue par un effet négatif de l’expansion polyQ sur l’interaction huntingtine (soluble)/HAP1. Dans des stades plus tardifs, les agrégats neuritiques s’accumulant pourraient également contribuer à la diminution du transport axonal (gain de fonction).
La huntingtine a des propriétés anti-apoptotiques
La huntingtine est une protéine enrichie dans le cerveau qui est essentielle au développement embryonnaire et à la neurogenèse [8]. La huntingtine sauvage, au contraire de la huntingtine polyQ, possède des propriétés anti-apoptotiques dans différents modèles cellulaires [3]. De plus, chez les souris mutantes pour la huntingtine, une augmentation de la mort cellulaire programmée est observée au cours du développement [9]. En accord avec ces données, les résultats de L.R. Gauthier et al. [2] démontrent que les propriétés anti-apoptotiques de la huntingtine sont liées, au moins en partie, à sa capacité de promouvoir le transport de BDNF dans le cerveau.
En situation polyQ, les propriétés anti-apoptotiques liées au transport vésiculaire sont perdues, mais peuvent être compensées par l’expression de la huntingtine sauvage et non de la huntingtine polyQ. Ainsi, l’approche de vidéomicroscopie rapide 3D devrait permettre non seulement de valider si des composés neuroprotecteurs dans la maladie de Huntington sont capables de corriger le dysfonctionnement de la huntingtine, mais également d’identifier de nouveaux composés à intérêt thérapeutique.
Références
- Ross CA. Polyglutamine pathogenesis : emergence of unifying mechanisms for Huntington’s disease and related disorders. Neuron 2002; 35 : 819–22. [Google Scholar]
- Gauthier LR, Charrin BC, Borell-Pagès M, et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 2004; 118 : 127–38. [Google Scholar]
- Cattaneo E, Rigamonti D, Goffredo D, et al. Loss of normal huntingtin function : New developments in Huntington’s disease research. Trends Neurosci 2001; 24 : 182–88. [Google Scholar]
- MacDonald ME, Gines S, Gusella JF, et al. Huntington’s disease. Neuromol Med 2003; 4 : 7–20. [Google Scholar]
- DiFiglia M. Huntingtin fragments that aggregate go their separate ways. Mol Cell 2002; 10 : 224. [Google Scholar]
- Saudou F, Finkbeiner S, Devys D, et al. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 1998; 95 : 55–66. [Google Scholar]
- Gunawardena S, Her LS, Brusch RG, et al. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron 2003; 40 : 25–40. [Google Scholar]
- White JK, Auerbach W, Duyao MP, et al. Huntingtin is required for neurogenesis and is not impaired by the Huntington’s disease CAG expansion. Nat Genet 1997; 17 : 404–10. [Google Scholar]
- Zeitlin S, Liu JP, Chapman DL, et al. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington’s disease gene homologue. Nat Genet 1995; 11 : 155–63. [Google Scholar]
© 2004 médecine/sciences - Inserm / SRMS
Liste des figures
|
Figure 1. La huntingtine et le BDNF sont colocalisés dans des cellules d’origine neuronale. Les cellules sont fixées, immunomarquées pour la huntingtine (en rouge) et pour le BDNF (brain-derived neurotrophic factor) (en vert), puis analysées par microscopie à déconvolution (photo, Jim P. Dompierre, Institut Curie, Orsay, France). |
| Dans le texte | |
|
Figure 2. Fonction de la huntingtine dans le transport du BDNF. A. La huntingtine normale s’associe au complexe dynéine-dynactine qui est un des moteurs moléculaires responsable du transport de cargos le long des microtubules. Cette association se fait via la protéine HAP1 (huntingtin-associated protein 1) : la huntingtine normale agit alors comme un facteur de processivité pour le transport des vésicules contenant BDNF.B. En situation pathologique, l’association huntingtine/HAP1/dynactine est altérée, ce qui a pour conséquence un détachement des protéines du complexe moteur des microtubules. Dans ces conditions, le transport de BDNF est réduit (schéma réalisé par Fabrice P. Cordelières, Institut Curie, Orsay, France). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.