")
")
| Issue |
Med Sci (Paris)
Volume 20, Number 6-7, Juin-Juillet 2004
|
|
|---|---|---|
| Page(s) | 643 - 650 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/2004206-7643 | |
| Published online | 15 juin 2004 | |
Remodelage précoce du ventricule gauche après un accident coronarien aigu
Early left ventricular remodelling following acute coronary syndromes
1
Inserm U. 460, Groupe Hospitalier Bichat-Claude Bernard, 46, rue Henri Huchard, 75018 Paris, France
2
Sevice de Cardiologie, Hôpital Beaujon, 100, boulevard du Général Leclerc, 92110 Clichy, France
3
Service de Physiologie-Explorations Fonctionnelles, Groupe Hospitalier Bichat-Claude Bernard
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Le remodelage ventriculaire survenant à la suite d’un syndrome coronarien aigu est complexe et multiforme. Il est le fait de la réponse du myocarde aux différentes agressions intervenant au cours de ces syndromes, principalement de l’ischémie et de la nécrose du territoire en aval de l’artère occluse. Il ne faut toutefois pas négliger la responsabilité des lésions liées à la reperfusion spontanée ou provoquée des tissus ou encore liées aux embolies dans la microcirculation coronaire de divers types de cellules et de débris responsables du phénomène d’absence de reperfusion (no-reflow). Le remodelage à la phase aiguë de l’infarctus est dominé par la dilatation précoce du ventricule qui conditionne largement le pronostic tardif, la détersion de la nécrose remplacée par une fibrose cicatricielle et l’hypertrophie compensatrice associée à la fibrose des zones non infarcies survenant parallèlement. Les mécanismes cellulaires et moléculaires de ces divers aspects du remodelage sont de mieux en mieux connus, permettant d’expliquer l’effet bénéfique de nombreuses molécules déjà disponibles et offrant aussi des cibles potentielles pour de nouvelles thérapeutiques. Une valorisation de ces connaissances associée à l’identification de facteurs de risque de remodelage et à une intervention thérapeutique toujours plus précoce devrait permettre de limiter encore plus les aspects délétères de ce processus, afin d’éviter ou retarder encore l’évolution vers l’insuffisance cardiaque.
Abstract
Ventricular remodelling following acute coronary syndromes is both complex and multiform. It is due to the response of the myocardium to the different agressions associated with these syndromes, in particular the ischemia and necrosis downstream of the occluded artery. We must not however neglect the role of the remodelling of the lesions resulting from spontaneous reperfusion or provoked by the cells and tissues associated with coronary microcirculation embolisms and the no-reflow phenomenon. Acute post-infarct remodelling is dominated by early ventricular dilatation which largely affects late prognosis, necrosis elimination and its replacement by a fibrotic scar in parallel with a compensatory hypertrophy of the non-infarcted myocardium. The diverse cellular and molecular components of this remodelling are increasingly well-known, allowing us to better explain the beneficial effects of the currently available medications and providing us with new potential therapeutic targets. A grading of this knowledge associated with the identification of new risk factors and early therapeutic interventions should help us to further limit the deleterious aspects of this remodelling in the goal of preventing, or at least delaying, the devolution towards heart failure.
© 2004 médecine/sciences - Inserm / SRMS
Le terme « remodelage » désigne une dynamique biologique qui se développe au sein d’un organe ou d’un tissu et qui, à travers des mécanismes à visée le plus souvent adaptative, conduit finalement à une dégradation de sa fonction. Ce processus est le fait de la plasticité du vivant qui permet aux cellules de modifier l’expression de leurs gènes en fonction de modifications de leur environnement. Celles-ci sont à l’origine de nombreux signaux qui sont intégrés par l’organe, le tissu et les cellules qui le constituent, ces dernières activant diverses voies de transduction conduisant à la reprogrammation de l’expression de leurs gènes. Ces cascades de signaux extra- et intracellulaires constituent autant de cibles potentielles pour des thérapeutiques visant à contrôler le remodelage et à en prévenir les aspects délétères.
Ce schéma général s’applique particulièrement bien aux événements biologiques survenant, au niveau du cœur, à la suite des multiples et diverses agressions que constituent pour le myocarde et les cellules qui le constituent les syndromes coronariens aigus (Encadré 1). Ceux-ci, au contraire de la brève ischémie myocardique de l’angor d’effort, dont l’effet de préconditionnement est protecteur, conduisent à divers degrés de lésion myocardique, qui vont des lésions topographiquement limitées, faisant suite à des emboles plaquettaires à partir d’un caillot formé sur une plaque rompue dans le cadre de l’angor instable, jusqu’à la nécrose des cellules de toute l’épaisseur - de l’endocarde à l’épicarde, l’infarctus est alors appelé transmural - d’un segment du ventricule gauche, à la suite de l’occlusion complète et prolongée d’une grosse artère épicardique.
Si les mécanismes du remodelage ventriculaire secondaire à un infarctus transmural sont maintenant bien connus, en particulier grâce à l’étude de modèles expérimentaux simples, il n’en va pas de même des remodelages succédant aux embolies distales spontanées (dans le cadre de l’angor instable) ou associées aux procédures de revascularisation, ou encore aux lésions infligées au myocarde par la reperfusion du tissu ischémique, qu’il s’agisse là encore d’un processus survenant lors de la lyse spontanée du caillot ou, au contraire, provoqué par les procédures de revascularisation. Au décours de l’infarctus transmural, le remodelage est caractérisé par la dilatation et la déformation progressive de la chambre ventriculaire, associée à une désadaptation du couplage ventriculo-artériel.
1. Types et stades de souffrance du myocarde à l’occasion des syndromes coronariens aigus et de leur traitement
-
L’angor d’effort résulte d’une ischémie transitoire du myocarde liée à un déséquilibre entre ses besoins en oxygène et les capacités du réseau coronaire à y répondre, en raison d’une réduction du calibre d’une ou de plusieurs artères par des plaques d’athérome : il cesse avec l’arrêt de l’effort. À ce stade, les plaques sont stables et ne font que rétrécir le calibre du vaisseau. Un ou plusieurs de ces épisodes « protègent » (relativement) le myocarde lors d’un infarctus ultérieur, celui-ci étant moins étendu que s’il n’avait pas été précédé par un tel épisode (notion de préconditionnement) (‹). La répétition de ces épisodes est associée à un remodelage moléculaire des myocytes (par exemple, expression de l’isoforme B de la créatine kinase).
(‹) m/s 2004, n° 5, p. 521
-
L’angor instable correspond à des douleurs qui ne sont, cette fois, plus provoquées par l’effort, mais spontanées. La rupture (dans 60 % à 70 % des cas) ou l’érosion (30 % à 40 % des cas) d’une plaque provoquent l’activation des plaquettes et de la coagulation, et la constitution d’un caillot (‹) qui n’est pas obligatoirement occlusif. Il peut alors envoyer dans la microcirculation coronaire d’aval des emboles fibrinoplaquettaires responsables d’une souffrance cellulaire.
(‹) ms 2004, n° 3, p. 293
Si le thrombus devient occlusif et qu’il ne se lyse pas de façon spontanée ou provoquée, l’ischémie myocardique perdure et conduit à l’infarctus du myocarde, qui associe divers degrés d’apoptose et de nécrose cellulaire.
Si le thrombus se lyse spontanément sous l’effet de l’activation de la fibrinolyse endogène ou à la suite d’une thrombolyse thérapeutique, la reperfusion du territoire ischémique (processus « séquence ischémie-reperfusion ») est responsable de lésions myocardiques spécifiques, liées notamment à un stress oxydatif intense et à une activation de l’échangeur Na+-H+. La sortie des ions H+ des myocytes contre une entrée de Na+ freine l’expulsion du Ca2+ à l’extérieur de la cellule par l’échangeur Na+-Ca2+ : l’augmentation du Ca2+ à l’intérieur des myocytes est responsable de divers types de dégâts cellulaires.
À l’occasion des manœuvres de recanalisation d’une artère occluse, le matériel constitutif du thrombus et divers débris peuvent migrer en aval et occlure la microcirculation coronaire, aboutissant au phénomène de non-reperfusion (no reflow). Ce phénomène est lui aussi responsable d’un stress spécifique pour la microcirculation et prolonge l’ischémie des myocytes.
Au total, un même territoire myocardique peut être victime séquentiellement de tous ces processus physiopathologiques combinant divers types/degrés de souffrance et de protection/sauvetage cellulaire. L’évaluation de l’importance relative et des conséquences myocardiques à long terme de chacun de ces processus reste à ce jour impossible.
Qu’est-ce que le remodelage ventriculaire post-infarctus ?
Le remodelage ventriculaire est un processus complexe, dynamique, qui débute dans les toutes premières minutes suivant l’occlusion coronaire et se poursuit pendant plusieurs semaines (Encadré 2). Chez le rat, où il a été étudié extensivement à la suite de la ligature de l’artère interventriculaire antérieure, on peut le décrire dans le temps selon trois phases et, dans l’espace, selon trois niveaux.
2. Problématique du remodelage myocardique après infarctus
Le temps 0 indique le moment de survenue de l’infarctus. Quand celui-ci est étendu, la phase aiguë (phase I) est caractérisée par une altération de la fonction ventriculaire gauche ayant pour conséquence une dilatation du ventricule ; celle-ci augmente alors les contraintes pariétales systoliques et diastoliques (augmentation du rayon r dans la formule sigma = kPr/e, où P représente la pression qui règne dans le ventricule et e l’épaisseur de sa paroi), ce qui entraîne un étirement des tissus nécrosés et non nécrosés. La baisse parallèle de la pression artérielle moyenne s’accompagne d’une activation des systèmes neuro-hormonaux (essentiellement catécholamines et système rénine-angiotensine-aldostérone), dont le but principal est de la maintenir à un niveau normal. Facteurs mécaniques et facteurs neuro-hormonaux déclenchent presque immédiatement le remodelage des régions nécrosées et non nécrosées. Si les processus adaptatifs l’emportent, l’activation neuro-hormonale décroît et l’on entre dans la phase II de remodelage compensé au cours de laquelle les contraintes sont plus ou moins normalisées ; cependant, le remodelage cardiaque persiste (ligne rouge pointillée). Si, au contraire, la dilatation ventriculaire et la baisse de la pression artérielle moyenne persistent, le passage s’effectue d’emblée dans la phase III d’insuffisance cardiaque chronique, caractérisée par la persistance de l’élévation des contraintes mécaniques, de l’activation des systèmes neuro-hormonaux et l’exacerbation du remodelage, l’ensemble conduisant à plus ou moins long terme au décès des patients. Toutes les interventions thérapeutiques de la phase aiguë des syndromes coronariens et les interventions thérapeutiques ultérieures, y compris les divers types de thérapie cellulaire à la phase aiguë ou dans les phases ultérieures, tentent de normaliser les contraintes mécaniques et de bloquer l’activation des systèmes neuro-hormonaux ou leurs effets délétères sur le myocarde, afin d’éviter l’entrée dans le cercle vicieux de la phase III où l’élévation des contraintes altère encore davantage la fonction ventriculaire, avec comme conséquence une majoration de l’activation des systèmes neuro-hormonaux, et donc une aggravation des contraintes mécaniques et des aspects délétères du remodelage.
Phases du remodelage post-infarctus
Les premiers jours associent nécrose cellulaire, œdème et inflammation tissulaire : les polynucléaires neutrophiles et les macrophages envahissent la zone infarcie et phagocytent les myocytes nécrosés jusqu’à la détersion complète du tissu infarci. La quantité de cellules inflammatoires se localisant principalement au niveau de la zone frontière entre tissu nécrosé et tissu sain croît jusqu’à la fin de la première semaine, puis décroît.
La seconde phase est la réparation fibrotique de la zone nécrosée : les myofibroblastes jouent un rôle essentiel dans ce processus, dès le troisième jour qui suit l’infarctus. Au 7e jour, on distingue trois zones dans l’infarctus : la zone centrale nécrosée, entourée d’une zone de macrophages et de myofibroblastes, puis d’une zone périphérique ou prédominent les myofibroblastes.
Un mois après l’infarctus, on ne retrouve pratiquement plus de cellules inflammatoires et les myofibroblastes ont envahi la totalité de la zone infarcie qui est le siège d’un important dépôt de collagène. Parallèlement, le myocarde non infarci subit un remodelage hypertrophique associant hypertrophie des myocytes, hyperplasie des cellules non musculaires et fibrose interstitielle.
Niveaux du remodelage post-infarctus
Le remodelage concerne les trois niveaux d’organisation structurale et fonctionnelle du cœur, auxquels on peut rajouter un 4e niveau moléculaire.
Le premier niveau est celui de la pompe cardiaque ou, pour simplifier, du ventricule gauche, structure tridimensionnelle de forme ellipsoïde constituée de myocarde réparti autour d’une cavité dont le volume change en permanence au cours du cycle cardiaque.
Le second niveau est celui du myocarde, tissu musculaire constitué de myocytes répartis en plusieurs couches, de cellules non musculaires, de vaisseaux et de matrice extracellulaire. Ce niveau inclut la zone infarcie et la zone non infarcie du ventricule. La région infarcie subit un remodelage cicatriciel, tandis que les régions du myocarde qui n’ont pas subi - ou ne vont pas subir pendant la phase aiguë - d’ischémie participeront, essentiellement par l’intermédiaire d’une hypertrophie des myocytes, au processus d’adaptation de la pompe ventriculaire à ses nouvelles conditions de fonctionnement.
Le troisième niveau est celui des cellules constitutives du myocarde, et en premier lieu des myocytes, cellules musculaires striées présentant de grandes similitudes structurales et fonctionnelles avec les cellules des muscles du squelette, qui vont accroître leur volume par divers degrés d’augmentation de longueur ou d’épaisseur selon le type de surcharge mécanique qu’ils subissent.
Le quatrième niveau est moléculaire, des isoformes de protéines absentes à l’état normal se trouvant anormalement exprimées (actine alpha-squelettique), d’autres voyant leur expression augmentée (chaîne lourde bêta de la myosine, peptides natriurétiques de type A et B…) ou encore diminuée (chaîne lourde alpha de la myosine, Ca2+-ATPase du réticulum sarcoplasmique…) à l’occasion du processus de remodelage (réexpression du programme fœtal).
Dans le post-infarctus, chacun de ces niveaux est concerné par un processus de remodelage spécifique et relativement autonome. Ils se retrouvent dans les autres situations pathologiques qui créent une surcharge chronique de travail hémodynamique pour le ventricule gauche, mais le cas de l’infarctus est compliqué par la coexistence de deux tissus dont l’évolution est distincte, voire opposée : la cicatrice, qui va devoir résister aux contraintes mécaniques, et les régions non nécrosées, qui vont participer aux mécanismes d’adaptation de la pompe ventriculaire à ses nouvelles conditions de fonctionnement.
La dilatation ventriculaire, primum movens du remodelage cardiaque
Dans le cas typique d’un infarctus transmural d’étendue significative, le remodelage du ventricule est caractérisé par une dilatation extrêmement précoce, observée dès la 3e heure et n’évoluant pratiquement plus pendant la première semaine [1].
La perte de fonction contractile d’une partie du myocarde amène en effet le ventricule à se dilater presque instantanément pour maintenir son volume d’éjection au prix d’une élévation de la pression qui y règne en fin de remplissage. Cette dilatation est adaptative, tendant à maintenir le débit cardiaque à la suite de la perte d’une partie du capital contractile. Par le mécanisme de Frank-Starling, l’étirement des myofibres permet en effet d’augmenter la contractilité des myocytes. En outre, un moindre raccourcissement des sarcomères est suffisant pour assurer un même débit cardiaque quand les dimensions du ventricule sont augmentées. Il s’y associe une déformation de la chambre ventriculaire dont l’importance dépend de la taille et de l’évolution de la zone infarcie. Avec la dilatation, la déformation de la chambre ventriculaire est responsable d’un découplage ventriculo-aortique, cause d’une augmentation de l’impédance systolique, véritable moteur de la surcharge mécanique chronique du ventricule. Il en résulte un déséquilibre entre l’élévation des contraintes mécaniques, qui s’exercent sur les régions indemnes et nécrosées du ventricule, et la capacité de chacune de ces régions à développer les forces nécessaires pour assurer un débit cardiaque suffisant.
L’augmentation du volume du ventricule, sa déformation, le découplage ventriculo-artériel en résultant et la dysfonction contractile qui y est associée, mis en évidence dès les premiers jours qui suivent l’infarctus [2], constituent l’élément central du remodelage précoce du post-infarctus et de la dégénérescence à plus long terme de la fonction pompe du cœur : c’est le plus fort facteur prédictif de mortalité et de morbidité au cours des mois et des années qui vont suivre [3, 4]. C’est pourquoi toutes les mesures thérapeutiques sont mises en œuvre pour éviter la dilatation en agissant sur ses causes le plus en amont possible : diminution de la durée et de l’étendue de l’ischémie myocardique par une revascularisation aussi rapide que possible ; diminution des lésions de reperfusion et, en cas d’infarctus constitué, limitation de la dilatation ventriculaire. Ces objectifs constituent autant de sujets de recherche expérimentale et clinique pour diminuer la mortalité, non plus à court terme, mais à moyen et long terme, des patients atteints d’un syndrome coronarien aigu.
Mécanismes impliqués dans le remodelage ventriculaire
L’élévation de la pression de remplissage du ventricule en fin de diastole est responsable d’un étirement des myocytes, principal mécanisme à l’origine de la reprogrammation de l’expression de leur génome qui conduit à leur hypertrophie et à l’augmentation de la masse du myocarde non nécrosé. Quand l’étirement diastolique persiste, il est responsable d’un allongement des myocytes par adjonction de sarcomères en série qui participe à la dilatation du ventricule. Mais, d’après la loi de Laplace (sigma = kP.r/e), à pression égale ou même faiblement diminuée, la dilatation du ventricule (augmentation du rayon r) est aussi responsable d’une augmentation de la charge (contrainte = sigma) et donc de la force que doivent développer les myocytes lorsqu’ils se contractent. Le mécanisme adaptatif mis en jeu est alors l’augmentation d’épaisseur (e) du myocarde qui tend à normaliser sigma. Globalement, l’élévation des contraintes systoliques et diastoliques exerce sur la zone infarcie en cours de cicatrisation des contraintes mécaniques importantes qui, à un moment où elle s’amincit par détersion de la nécrose, peut conduire à la constitution d’un anévrisme, voire à la rupture du ventricule [5, 6]. Dans le cercle vicieux de la dilatation du ventricule, l’hypertrophie du myocarde non nécrosé « court » en quelque sorte derrière la dilatation sans arriver à la « rattraper » [7], d’autant que l’hypertrophie myocardique ne résulte pratiquement pas de l’hyperplasie des myocytes, mais de leur hypertrophie qui, bien que potentiellement importante, n’est pas illimitée.
Deux grandes voies de signalisation sont activées par l’étirement des myocytes et la stimulation neuro-hormonale observée à la phase aiguë des infarctus de grande taille compliqués d’insuffisance ventriculaire gauche : la voie des MAP kinases, activées par la stimulation des récepteurs à 7 domaines transmembranaires couplés aux protéines G (essentiellement Galphas et Galphaq/alpha11 couplées aux récepteurs de la noradrénaline, de l’angiotensine II, et de l’endothéline) [8], et la voie de la calcineurine, activée par l’augmentation de la concentration en Ca2+ libre à l’intérieur des myocytes [9]. Ces deux voies aboutissent à l’activation de facteurs de transcription qui déclenchent, de façon additive ou non selon le gène considéré, le processus hypertrophique et la réexpression du programme fœtal, mais qui peuvent aussi favoriser l’apoptose des myocytes. Une troisième voie, de découverte récente [10], est également activée par l’étirement des myocytes : elle fait intervenir des signaux qui trouvent leur origine au niveau de la ligne Z du sarcomère et des protéines d’ancrage de la ligne Z à la membrane cellulaire (costamère), et sont transmis au noyaux par une cascade de protéines en cours d’identification, dont, peut-être, la T-cap/téléthonine, la calsarcine et la calcineurine.
Remodelage de la matrice extracellulaire
Dans les premiers jours qui suivent l’infarctus, la nécrose des myocytes et leur détersion par les cellules inflammatoires prédominent. Ces phénomènes impliquent la dégradation de la matrice extracellulaire (MEC) et la mise en place d’une fibrose substitutive. Le remodelage de la MEC est régulé en grande partie par les métalloprotéinases de la matrice (MMP). Les MMP sont divisées en six familles selon leur structure et leur spécificité de substrat. Elles sont caractérisées par une région de liaison au zinc et la présence d’un domaine catalytique. La partie carboxyterminale est responsable de la reconnaissance des substrats et de l’association avec leurs inhibiteurs (TIMP, tissue inhibitors of metalloproteinases). L’activité des MMP est régulée à trois niveaux : l’expression, l’activation, et l’inhibition [11] (Figure 1).
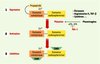 |
Figure 1. L’activité MMP tissulaire est contrôlée à trois niveaux. L’expression des MMP (métalloprotéases matricielles) est stimulée par l’étirement, ainsi que par des peptides tels que l’angiotensine II, le TGF-bêta (tumor necrosis factor-bêta) ou certaines cytokines. Elles sont sécrétées sous forme de zymogène et doivent être activées. Cette activation se fait par d’autres MMP, ou par des sérine-protéases telles que la plasmine. L’activité protéolytique des MMP peut être inhibée par des TIMP (tissue inhibitors of metalloproteinases). |
La dégradation précoce de la matrice par les MMP permet l’infiltration des cellules inflammatoires et des macrophages nécessaire à la détersion du tissu nécrotique et à la cicatrisation [12]. La lymphe cardiaque post-ischémique est capable de stimuler la sécrétion et l’activation de la MMP-9 par les polynucléaires neutrophiles [13]. Etoh et al. [14] ont montré une activité gélatinolytique in vivo augmentée 80 minutes après l’infarctus. Bien que nécessaire pour la phagocytose des myocytes nécrosés, la dégradation précoce de la MEC est associée au risque de rupture du ventricule, puisque l’incidence de mortalité précoce par rupture ventriculaire est diminuée chez les souris MMP-9−/− ou MMP-2−/− [12, 15].
Les MMP sont sécrétées par la cellule sous forme de zymogènes et nécessitent une activation par clivage protéolytique. In vivo, cette activation est réalisée par d’autres MMP ou par des sérine-protéases. Le système plaminogène/plasmine est impliqué dans cette activation (pour revue, voir [16]). Le plasminogène circulant est activé en plasmine par d’autres sérine-protéases, l’uPA (urokinase-type plasminogen activator) et le tPA (tissue-type plasminogen activator), ces activateurs étant inhibés par le PAI-1. L’importance de ce système est démontrée dans le modèle d’infarctus constitué chez la souris dont le gène du plasminogènea été invalidé [17]. Dans ce modèle, les cellules inflammatoires ne migrent pas dans la zone infarcie et on observe la persistance d’une zone nécrotique plusieurs semaines après l’infarctus. La dilatation cavitaire n’est cependant pas prévenue. Plus en aval, les souris uPA−/− montrent également une altération de la cicatrisation normale et une persistance de la zone nécrotique. Une surexpression transitoire de PAI-1 donne des résultats semblables. La cicatrisation tardive chez ces souris est associée à une baisse du risque de rupture cavitaire [12].
Les TIMP, inhibiteurs endogènes des MMP, s’y associent par une liaison non covalente dans un ratio stœchiométrique de 1 :1. Au nombre de quatre (TIMP-1 à -4), seule la TIMP-4 est plus ou moins spécifique du cœur [18]. Les données concernant l’expression des TIMP dans le post-infarctus sont disparates : on observe généralement des augmentations de l’expression de leurs ARNm [19–22], mais les résultats divergent au niveau protéique [23–26]. Cependant, une baisse des taux protéiques des TIMP est le plus souvent rapportée, qui reflète une régulation post-transcriptionnelle importante. Cette baisse des TIMP implique un environnement plus permissif à la dégradation de la MEC par les MMP.
Le revers de la médaille de la dégradation de la MEC est son dépôt. La fibrose substitutive tente de maintenir l’intégrité structurale de la zone infarcie à la suite de la nécrose et de la détersion des myocytes. Les myofibroblastes apparaissent dans la zone infarcie dès le 3e jour et se colocalisent avec l’expression des collagènes I et III [22]. Les taux de collagène sont augmentés surtout dans la zone infarcie, mais aussi dans le myocarde non infarci. On observe dès le 3e jour après l’infarctus une augmentation des ARNm, avec un pic au 7e jour et une diminution ensuite [22, 27]. L’accumulation de collagène est donc plus tardive que sa dégradation, le décalage représentant la période de risque de rupture du ventricule. Les efforts thérapeutiques devraient logiquement viser à réduire une dégradation trop rapide de la MEC dans les premiers jours post-infarctus. Cependant, cette dégradation est nécessaire à la cicatrisation, comme le démontre les travaux d’Heymans et al. et de Creemers et al. menés chez des souris génétiquement modifiées [12, 17], et il est donc capital de ne pas la bloquer entièrement. L’accumulation chronique de collagène dans les semaines et les mois qui suivent l’infarctus représente cependant une menace importante pour la fonction ventriculaire [28], mais qui est maintenant minimisée par l’utilisation thérapeutique des inhibiteurs du système rénine-angiotensine-aldostérone.
Reperfusion coronaire et phénomène d’absence de reperfusion (no-reflow)
L’étendue de l’infarctus et son caractère transmural sont des déterminants essentiels du remodelage ventriculaire [29, 30]. La réouverture précoce de l’artère occluse et la reperfusion tissulaire qui en découle limitent la taille de la zone infarcie et s’accompagnent d’un bénéfice clinique net en termes de mortalité, de survenue d’insuffisance cardiaque et d’arythmies [31]. Toutefois, une proportion de patients semble ne pas tirer bénéfice de cette reperfusion. Les dommages causés par la reperfusion sont connus [32] et incluent notamment des lésions au niveau de la microvascularisation. Ces lésions associent un dysfonctionnement endothélial à une obstruction microvasculaire, liée à une embolisation distale de débris thrombotiques et d’érythrocytes, ainsi qu’à la migration de polynucléaires. Il s’agit d’un phénomène dynamique prenant place au cours des premiers jours de l’infarctus. La conséquence en est finalement une insuffisance ou une absence de reperfusion (no-reflow) au niveau tissulaire, en dépit d’une artère épicardique réouverte [33], qui prolongent et aggravent l’ischémie et la mort cellulaire du territoire concerné. Différentes techniques cliniques permettent de documenter ce phénomène : visualisation échographique ou par IRM (imagerie par résonance magnétique) d’un défaut de prise de contraste tissulaire après injection intracoronaire ou périphérique d’un soluté, mesure de la réserve coronaire par Doppler.
La présence d’un phénomène de non-reperfusion a été associée au remodelage ventriculaire [34, 35]. De façon intéressante, la corrélation entre le degré d’obstruction microvasculaire et le remodelage semble indépendante de la taille de l’infarctus [36]. Cela semble être dû à un effet de l’obstruction microvasculaire sur la mécanique ventriculaire : en effet, l’obstruction microvasculaire altère très précocement la distensibilité de la zone infarcie (rôle de l’hémorragie et de l’œdème intramyocardique), puis des régions adjacentes [36], provoquant une augmentation du stress pariétal local et finalement du remodelage. Ainsi, un des défis thérapeutiques actuellement non résolu est l’obtention la plus rapide possible d’une reperfusion tissulaire optimale, et pas seulement la réouverture de l’artère épicardique.
Impact des thérapeutiques sur le remodelage ventriculaire post-infarctus
Traitements médicamenteux « antiremodelage »
De nombreux agents thérapeutiques visant à réduire le remodelage ventriculaire après l’infarctus sont couramment utilisés en clinique, en général après avoir été testés dans des modèles expérimentaux animaux, tandis que d’autres sont encore en cours d’étude dans ces modèles. Les dérivés nitrés donneurs de monoxyde d’azote (NO), par leurs effets vasodilatateurs artériels et veineux, réduisent les pressions artérielles et la pression de remplissage du ventricule, et donc les contraintes pariétales systoliques et diastoliques ; un effet favorable sur le remodelage et notamment sur la taille de l’infarctus a ainsi pu être observé. Toutefois, les études cliniques de mortalité réalisées chez des patients bénéficiant d’une reperfusion ont échoué à démontrer un quelconque bénéfice de ces agents [37, 38].
En revanche, les bêta-bloquants et les antagonistes du système rénine-angiotensine-aldostérone ont clairement montré un bénéfice sur la mortalité. Les bêta-bloquants agissent classiquement par le biais d’une baisse de la consommation en oxygène du myocarde, résultant en une réduction de la taille de l’infarctus et une diminution du risque d’arythmies. Il agissent aussi probablement en bloquant l’effet direct des catécholamines sur les myocytes. L’utilisation des inhibiteurs de l’enzyme de conversion de l’angiotensine I (IEC) a été validée par 7 essais randomisés, menés contre placebo, qui ont montré de façon univoque une réduction de la mortalité [39]. Dans plusieurs de ces essais, le traitement, débuté dans les 24 premières heures suivant l’infarctus, montrait un bénéfice dès le premier jour. Un effet hypotenseur précoce trop marqué s’est toutefois révélé délétère.
Comme pour l’insuffisance cardiaque chronique, le bénéfice obtenu avec ces agents dépasse celui attendu d’un simple effet vasodilatateur, suggérant une action spécifiquement liée à leur inhibition des effets cardiaques de l’activation du système rénine-angiotensine-aldostérone. En effet, le remodelage est, au-delà des catécholamines, largement influencé par l’angiotensine II et l’aldostérone [40, 41]. De plus, ces agents diminuent le renouvellement (turn-over) du collagène dans divers modèles expérimentaux. Plusieurs essais cliniques ont montré qu’un antagoniste de l’aldostérone prescrit à faible dose (et donc dénué d’effet hypotenseur) diminuait la mortalité post-infarctus. De façon intéressante, cet effet était plus marqué chez les patients présentant des stigmates biologiques sanguins suggérant un turn-over plus important de la matrice extracellulaire (présence de fragments de dégradation du collagène ou du fragment aminoterminal du procollagène de type III) [42]. L’éplerone, un antagoniste de l’aldostérone de ce type, prescrit à plus de 6 000 patients entre les 3e et 14e jours après l’infarctus, a réduit la mortalité de 15 % [43]. Un autre essai a évalué plus précisément l’effet de la spironolactone, prescrite en plus des IEC dans les premières heures d’un infarctus antérieur reperfusé [44] : les patients traités avaient un mois après l’infarctus un ventricule nettement moins dilaté que ceux du groupe témoin et une fraction d’éjection du ventricule supérieure. L’hypothèse d’une action suppressive de la spironolactone sur le turn-over du collagène était à nouveau confortée par une réduction de la concentration plasmatique de procollagène de type III.
Une action plus directe sur la matrice extracellulaire (MEC) peut être obtenue avec des inhibiteurs des métalloprotéases matricielles (MMP) ou de leurs inhibiteurs endogènes (TIMP). L’utilisation de certains inhibiteurs des MMP résulte en une moindre dilatation ventriculaire dans l’insuffisance cardiaque expérimentale [45–47]. Toutefois, une transposition de ces résultats au contexte de l’infarctus aigu n’est pas évidente en raison d’un rôle différent du remodelage de la matrice extracellulaire, au cours du temps, dans les zones infarcie et non infarcie. En effet, de tels agents peuvent exposer à une moindre cicatrisation de la zone infarcie, et donc à un plus haut risque de rupture ventriculaire.
Place potentielle d’une thérapie cellulaire
La recolonisation de la zone infarcie par des myocytes cardiaques est une stratégie thérapeutique en cours d’évaluation (‹).
(‹) m/s 2004, n° 6-7, p. 651
L’injection expérimentale, en zone infarcie ou en zone bordante de l’infarctus, de myoblastes ou de cellules pluripotentes ou progénitrices issues du sang ou de la moelle osseuse s’accompagne d’une amélioration du remodelage et de la fonction ventriculaire, et d’une néovascularisation. Néanmoins, la plupart de ces expériences ont été réalisées sur des infarctus constitués. Plusieurs essais cliniques ont déjà montré la faisabilité d’une telle pratique chez l’homme, par délivrance chirurgicale [48] ou endocoronaire [49] des cellules à distance de la phase aiguë. Les cellules de moelle osseuse semblent mieux adaptées pour recoloniser un infarctus aigu, avant la formation de la cicatrice fibreuse. Une étude menée sur 20 patients a également montré la faisabilité et la bonne tolérance de l’injection endocoronaire de telles cellules, précocement (3-4 jours) après un infarctus antérieur aigu reperfusé [50]. Ces résultats suggèrent une moindre dilatation ventriculaire, ainsi qu’une amélioration de la viabilité de la zone infarcie recolonisée par ces cellules ; toutefois, encore très préliminaires, ces données demandent à être validées par de grands essais multicentriques.
Conclusions
Les syndromes coronariens aigus doivent être considérés comme des modifications majeures de l’environnement cardiaque constituant autant de stress auxquels sont confrontés les myocytes, le myocarde et le ventricule. Face à ces stress, divers processus visant à préserver la fonction de l’organe sont mis en jeu, processus comportant souvent des aspects délétères à plus ou moins long terme, tels que la fibrose des régions non infarcies. De nombreux progrès thérapeutiques ont été réalisés au cours des dernières décennies pour minimiser ces effets délétères.
Il existe cependant des processus de défense naturelle du myocarde encore insuffisamment exploités au plan thérapeutique, tels que les mécanismes de pré- et post-conditionnement, ou encore la stimulation des voies de survie des myocytes lors des procédures de revascularisation myocardique. Ces mécanismes de mieux en mieux connus (‹) seront à n’en pas douter exploités au cours des années à venir pour atténuer le remodelage cardiaque consécutif aux accidents coronariens aigus.
(‹) m/s 2004, n° 5, p. 521
Enfin, il est certain que les capacités d’adaptation de chacun face à ces stress sont différentes. Des profils de patients à risque de remodelage délétère devraient pouvoir être établis à partir de phénotypes intermédiaires ou du génotypage des patients, autorisant en cela la mise en œuvre de procédures thérapeutiques spécifiques à chaque individu et à chaque statut coronarien.
Références
- Korup E, Dalsgaard D, Nyvad O, et al. Comparison of degrees of left ventricular dilation within three hours and up to six days after onset of first acute myocardial infarction. Am J Cardiol 1997; 80 : 449–53 [Google Scholar]
- Seals AA, Pratt CM, Mahmarian JJ, et al. Relation of left ventricular dilation during acute myocardial infarction to systolic performance, diastolic dysfunction, infarct size and location. Am J Cardiol 1988; 61 : 224–9. [Google Scholar]
- de Kam PJ, Nicolosi GL, Voors AA, et al. Prediction of 6 months left ventricular dilatation after myocardial infarction in relation to cardiac morbidity and mortality. Application of a new dilatation model to GISSI-3 data. Eur Heart J 2002; 23 : 536–42. [Google Scholar]
- White HD, Norris RM, Brown MA, et al. Left ventricular end-systolic volume as the major determinant of survival after recovery from myocardial infarction. Circulation 1987; 76 : 44–51. [Google Scholar]
- Weber KT, Sun Y, Tyagi SC, Cleutjens JP. Collagen network of the myocardium : Function, structural remodeling and regulatory mechanisms. J Mol Cell Cardiol 1994; 26 : 279–92. [Google Scholar]
- Gao XM, Dart AM, Dewar E, et al. Serial echocardiographic assessment of left ventricular dimensions and function after myocardial infarction in mice. Cardiovasc Res 2000; 45 : 330–8. [Google Scholar]
- Pfeffer JM, Pfeffer MA, Fletcher PJ, Braunwald E. Progressive ventricular remodeling in rat with myocardial infarction. Am J Physiol 1991; 260 : H1406–14. [Google Scholar]
- Sugden PH. Ras, Akt, and mechanotransduction in the cardiac myocyte. Circ Res 2003; 93 : 1179–92. [Google Scholar]
- Wilkins BJ, Molkentin JD. Calcineurin and cardiac hypertrophy : Where have we been ? Where are we going ? J Physiol (Lond) 2002; 541 : 1–8. [Google Scholar]
- Knöll R, Hoshijima M, Hoffmann HM, et al. The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell 2002; 111 : 943–55. [Google Scholar]
- Nagase H, Woessner JF Jr. Matrix metalloproteinases. J Biol Chem 1998; 274 : 21491–4. [Google Scholar]
- Heymans S, Luttun A, Nuyens D, et al. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat Med 1999; 5 : 1135–42. [Google Scholar]
- Lindsey M, Wedin K, Brown MD, et al. Matrix-dependent mechanism of neutrophil-mediated release and activation of matrix metalloproteinase 9 in myocardial ischemia/reperfusion. Circulation 2001; 103 : 2181–7. [Google Scholar]
- Etoh T, Joffs C, Deschamps AM, et al. Myocardial and interstitial matrix metalloproteinase activity after acute myocardial infarction in pigs. Am J Physiol Heart Circ Physiol 2001; 281 : H987–94. [Google Scholar]
- Hayashidani S, Tsutsui H, Ikeuchi M, et al. Targeted deletion of MMP-2 attenuates early LV rupture and late remodeling after experimental myocardial infarction. Am J Physiol Heart Circ Physiol 2003; 285 : H1229–35. [Google Scholar]
- Lijnen H. R. Plasmin and matrix metalloproteinases in vascular remodeling. Thromb Haemost 2001; 86 : 324–33. [Google Scholar]
- Creemers E, Cleutjens J, Smits J, et al. Disruption of the plasminogen gene in mice abolishes wound healing after myocardial infarction. Am J Pathol 2000; 156 : 1865–73. [Google Scholar]
- Rahkonen OP, Koskivirta IM, Oksjoki SM, et al. Characterization of the murine Timp4 gene, localization within intron 5 of the synapsin 2 gene and tissue distribution of the mRNA. Biochim Biophys Acta 2002; 1577 : 45–52. [Google Scholar]
- Cleutjens JP, Kandala JC, Guarda E, et al. Regulation of collagen degradation in the rat myocardium after infarction. J Mol Cell Cardiol 1995; 27 : 1281–92. [Google Scholar]
- Tyagi SC, Kumar SG, Haas SJ, et al. Post-transcriptional regulation of extracellular matrix metalloproteinase in human heart end-stage failure secondary to ischemic cardiomyopathy. J Mol Cell Cardiol 1996; 28 : 1415–28. [Google Scholar]
- Peterson JT, Li H, Dillon L, Bryant JW. Evolution of matrix metalloprotease and tissue inhibitor expression during heart failure progression in the infarcted rat. Cardiovasc Res 2000; 46 : 307–15. [Google Scholar]
- Sun Y, Zhang JQ, Zhang J, Lamparter S. Cardiac remodeling by fibrous tissue after infarction in rats. J Lab Clin Med 2000; 135 : 316–23. [Google Scholar]
- Li YY, Feldman AM, Sun Y, McTierman CF. Differential expression of tissue inhibitors of metalloproteinases in the failing human heart. Circulation 1998; 98 : 1728–34. [Google Scholar]
- Spinale FG, Coker ML, Heung LJ, et al. A matrix metalloproteinase induction/activation system exists in the human left ventricular myocardium and is upregulated in heart failure. Circulation 2000; 102 : 1944–9. [Google Scholar]
- Romanic AM, Burns-Kurtis CL, Gout B, et al. Matrix metalloproteinase expression in cardiac myocytes following myocardial infarction in the rabbit. Life Sci 2001; 68 : 799–814. [Google Scholar]
- Fedak PW, Altamentova SM, Weisel RD, et al. Matrix remodeling in experimental and human heart failure : A possible regulatory role for TIMP-3. Am J Physiol Heart Circ Physiol 2003; 284 : H626–34. [Google Scholar]
- Zimmerman SD, Thomas DP, Velleman SG, et al. Time course of collagen and decorin changes in rat cardiac and skeletal muscle post-MI. Am J Physiol Heart Circ Physiol 2001; 281 : H1816–22. [Google Scholar]
- Beltrami CA, Finato N, Rocco M, et al. Structural basis of end-stage failure in ischemic cardiomyopathy in humans. Circulation 1994; 89 : 151–63. [Google Scholar]
- Hochman JS, Choo H. Limitation of myocardial infarct expansion by reperfusion independent of myocardial salvage. Circulation 1987; 75 : 299–306. [Google Scholar]
- Chareonthaitawee P, Christian TF, Hirose K, et al. Relation of initial infarct size to extent of left ventricular remodeling in the year after acute myocardial infarction. J Am Coll Cardiol 1995; 25 : 567–73. [Google Scholar]
- Lange RA, Hillis LD. Reperfusion therapy in acute myocardial infarction. N Engl J Med 2002; 346 : 954–5. [Google Scholar]
- Kloner RA. Does reperfusion injury exist in humans ? J Am Coll Cardiol 1993; 21 : 537–45. [Google Scholar]
- Ito H, Tomooka T, Sakai N, et al. Lack of myocardial perfusion immediately after successful thrombolysis. A predictor of poor recovery of left ventricular function in anterior myocardial infarction. Circulation 1992; 85 : 1699–705. [Google Scholar]
- Ito H, Maruyama A, Iwakura K, et al. Clinical implications of the ’no reflow’ phenomenon. A predictor of complications and left ventricular remodeling in reperfused anterior wall myocardial infarction. Circulation 1996; 93 : 223–8. [Google Scholar]
- Wu KC, Zerhouni EA, Judd RM, et al. Prognostic significance of microvascular obstruction by magnetic resonance imaging in patients with acute myocardial infarction. Circulation 1998; 97 : 765–72. [Google Scholar]
- Gerber BL, Rochitte CE, Melin JA, et al. Microvascular obstruction and left ventricular remodeling early after acute myocardial infarction. Circulation 2000; 101 : 2734–41. [Google Scholar]
- GISSI-3 trial. Six-month effects of early treatment with lisinopril and transdermal glyceryl trinitrate singly and together withdrawn six weeks after acute myocardial infarction : The GISSI-3 trial. Gruppo italiano per lo studio della sopravvivenza nell’infarto miocardico. J Am Coll Cardiol 1996; 27 : 337–44. [Google Scholar]
- ISIS-4 (Fourth International study of infarct survival) Collaborative Group. ISIS-4 : A randomised factorial trial assessing early oral captopril, oral mononitrate, and intravenous magnesium sulphate in 58,050 patients with suspected acute myocardial infarction. Lancet 1995; 345 : 669–85. [Google Scholar]
- Latini R, Maggioni AP, Flather M, et al. ACE inhibitor use in patients with myocardial infarction. Summary of evidence from clinical trials. Circulation 1995; 92 : 3132–7. [Google Scholar]
- Rouleau JL, de Champlain J, Klein M, et al. Activation of neurohumoral systems in postinfarction left ventricular dysfunction. J Am Coll Cardiol 1993; 22 : 390–8. [Google Scholar]
- Brilla CG, Zhou G, Matsubara L, Weber KT. Collagen metabolism in cultured adult rat cardiac fibroblasts : Response to angiotensin II and aldosterone. J Mol Cell Cardiol 1994; 26 : 809–20. [Google Scholar]
- Zannad F, Alla F, Dousset B, et al. Limitation of excessive extracellular matrix turnover may contribute to survival benefit of spironolactone therapy in patients with congestive heart failure : Insights from the randomized aldactone evaluation study (RALES). Rales Investigators. Circulation 2000; 102 : 2700–6. [Google Scholar]
- Pitt B, Remme W, Zannad F, et al. Eplerenone post-acute myocardial infarction heart failure efficacy and survival study investigators. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med 2003; 348 : 1309–21. [Google Scholar]
- Hayashi M, Tsutamoto T, Wada A, et al. Immediate administration of mineralocorticoid receptor antagonist spironolactone prevents post-infarct left ventricular remodeling associated with suppression of a marker of myocardial collagen synthesis in patients with first anterior acute myocardial infarction. Circulation 2003; 107 : 2559–65. [Google Scholar]
- Spinale FG, Coker ML, Krombach SR, et al. Matrix metalloproteinase inhibition during the development of congestive heart failure : Effects on left ventricular dimensions and function. Circ Res 1999; 85 : 364–76. [Google Scholar]
- Mukherjee R, Brinsa TA, Dowdy KB, et al. Myocardial infarct expansion and matrix metalloproteinase inhibition. Circulation 2003; 107 : 618–25. [Google Scholar]
- Yarbrough WM, Mukherjee R, Escobar GP, et al. Selective targeting and timing of matrix metalloproteinase inhibition in post-myocardial infarction remodeling. Circulation 2003; 108 : 1753–9. [Google Scholar]
- Menasche P, Hagege AA, Scorsin M, et al. Myoblast transplantation for heart failure. Lancet 2001; 357 : 279–80. [Google Scholar]
- Strauer BE, Brehm M, Zeus T, et al. Repair of infarcted myocardium by autologous intracoronary mononuclear bone marrow cell transplantation in humans. Circulation 2002; 106 : 1913–8. [Google Scholar]
- Assmus B, Schachinger V, Teupe C, et al. Transplantation of progenitor cells and regeneration enhancement in acute myocardial infarction (TOPCARE-AMI). Circulation 2002; 106 : 3009–17 [Google Scholar]
Liste des figures
|
Figure 1. L’activité MMP tissulaire est contrôlée à trois niveaux. L’expression des MMP (métalloprotéases matricielles) est stimulée par l’étirement, ainsi que par des peptides tels que l’angiotensine II, le TGF-bêta (tumor necrosis factor-bêta) ou certaines cytokines. Elles sont sécrétées sous forme de zymogène et doivent être activées. Cette activation se fait par d’autres MMP, ou par des sérine-protéases telles que la plasmine. L’activité protéolytique des MMP peut être inhibée par des TIMP (tissue inhibitors of metalloproteinases). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.