")
")
| Issue |
Med Sci (Paris)
Volume 20, Number 1, Janvier 2004
|
|
|---|---|---|
| Page(s) | 55 - 60 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/200420155 | |
| Published online | 15 January 2004 | |
Le système MMP/TIMP dans le système nerveux
The MMP/TIMP system in the pathophysiology of the nervous system
Neurobiologie des Interactions Cellulaires et Neurophysiopathologie (NICN), FRE Cnrs 2533, Université de la Méditerranée, Faculté de Médecine de Marseille, IFR Jean Roche, boulevard Pierre Dramard, 13916 Marseille Cedex 20, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
Les métalloprotéases matricielles (MMP) contrôlent ou dégradent par clivage protéolytique des composants de la matrice extracellulaire, des protéines d’adhérence, des récepteurs membranaires et des protéines solubles. Le contrôle de l’activité des MMP par leurs inhibiteurs physiologiques, les TIMP (tissue inhibitors of metalloproteinases), contribue à l’homéostasie tissulaire. En revanche, la perte de ce contrôle peut être associée à des processus de prolifération ou de mort cellulaire et aux remaniements tissulaires qui caractérisent les maladies malignes et dégénératives de nombreux tissus n’appartenant pas au système nerveux. Cet article fait le point sur les résultats récents montrant que les MMP jouent également un rôle central dans la physiopathologie du système nerveux et qu’elles pourraient constituer de nouvelles cibles thérapeutiques dans différentes maladies du système nerveux.
Abstract
The matrix metalloproteinases (MMP) belong to a growing family of secreted or membrane-bound (MT-MMP) enzymes that cleave protein components of the extracellular matrix and bioactive factors involved in intercellular signaling. MMP activity is counterbalanced by their four physiological inhibitors, the tissue inhibitors of MMP (TIMPs). Together, MMP and TIMP control cell-cell and cell-matrix interactions associated with physiological processes. However, the breakdown of the protease-inhibitor balance may lead to the loss of tissue homeostasis and the development of degenerative and tumorigenic processes in various tissues. The emerging idea is that the MMP/TIMP system also plays a major role in the pathology and physiology of the nervous system and that mastering MMP activity will set the basis for new and more efficient therapeutic strategies against nervous system disorders.
© 2004 médecine/sciences - Inserm / SRMS
Les métalloprotéases matricielles (MMP) constituent une famille multigénique (près de 25 membres à ce jour) de protéases dépendantes du zinc, sécrétées ou membranaires (membrane type MMP, MT-MMP). Les MMP contrôlent par clivage protéolytique l’activité de composants de la matrice extracellulaire, des molécules membranaires ou solubles impliquées dans la transmission des signaux intercellulaires telles que les cytokines, les chimiokines, les facteurs trophiques, les protéines d’adhérence et différents récepteurs. Dans les tissus, l’activité protéolytique des MMP est contrôlée par quatre inhibiteurs de MMP, les TIMP (tissue inhibitors of metalloproteinases) qui possèdent également des propriétés trophiques ou, à l’inverse, pro-apoptotiques. Le système MMP/TIMP contrôle les interactions cellule-cellule et cellule-matrice impliquées dans de nombreux processus physiologiques, notamment la prolifération, la différenciation, la migration et la mort cellulaire. Cependant, la rupture de l’équilibre protéase-inhibiteur peut entraîner dans de nombreux tissus la perte de l’homéostasie et le développement de processus dégénératifs ou cancéreux, en particulier les métastases (→). Si le système MMP/TIMP est très étudié en dehors du système nerveux, il ne suscite l’intérêt des neurobiologistes que depuis une dizaine d’années. Nous résumons ici les données récentes qui montrent que le système MMP/TIMP joue un rôle majeur dans différents aspects de la physiologie et de la pathologie du système nerveux et que les MMP pourraient constituer de nouvelles cibles thérapeutiques dans différentes neuropathies.
(→) m/s 2002, n°5, p. 565
Le système MMP/TIMP au cours de l’ontogenèse du système nerveux et dans la plasticité neuronale
La distribution spatio-temporelle de nombreuses MMP et des quatre TIMP varie selon le stade de développement considéré. Ainsi, les variations d’expression de TIMP-1, -2 et -3 et de MMP-2, -3 et -9 sont étroitement liées aux étapes successives du développement post-natal du cervelet [1]. L’expression de MT5-MMP, une MMP membranaire essentiellement exprimée dans le cerveau, varie au cours de l’ontogenèse, avec un pic à la naissance. Cette expression reste élevée dans des structures considérées comme plastiques (cervelet, hippocampe et bulbe olfactif), suggérant également une implication de MT5-MMP dans les processus physiologiques de plasticité neuronale [2]. La première allusion à une contribution du système MMP/TIMP dans la plasticité neuronale remonte à 1993, lorsque Y. Citri et son équipe ont montré une forte induction de TIMP-1 dans les neurones des grains du gyrus denté au cours de la potentialisation à long terme, un modèle d’étude de la plasticité (pour revue, voir [3]). Le gène timp-1 a alors été inclus parmi les gènes potentiellement impliqués dans la plasticité (candidate plasticity gene). La plasticité neuronale se caractérise, entre autres, par des processus de croissance des neurites et quelques données permettent déjà d’impliquer le système MMP/TIMP dans l’axogenèse : sur des préparations de moelle épinière, des inhibiteurs de métalloprotéases inhibent la protéolyse de DCC (deleted in colorectal cancer), un récepteur de la nétrine-1, facteur permettant le guidage des axones (→). Il en résulte une potentialisation des effets de la nétrine-1 et il a été proposé que l’activité des métalloprotéases pourrait contrôler la migration axonale en modulant le nombre de récepteurs de la nétrine-1 à la surface des axones [4]. La croissance axonale est potentialisée par différents facteurs neurotrophiques mais il reste beaucoup à apprendre quant à leurs « effecteurs ». Or il a été montré sur des neurones des ganglions dorsaux que le NGF (nerve growth factor) induit la synthèse de MMP-2 : la stimulation de l’axogenèse par le NGF est liée à la dégradation par MMP-2 de protéoglycanes à chondroïtine sulfate (CSPG) de la matrice extracellulaire qui inhibent la croissance neuritique (pour revue, voir [3]). Plus récemment, il a été montré in vivo que les inhibiteurs de MMP perturbent la progression et le guidage des axones de la rétine chez le xénope [5]. L’implication des métalloprotéases dans l’axogenèse et dans le guidage axonal est étayée par la conservation phylogénique des mécanismes mis en jeu et des systèmes protéiques cibles de ces protéases. Ainsi, Dm1-MMP, l’une des deux MMP décrites à ce jour chez Drosophila melanogaster, est spécifiquement exprimée dans les cellules gliales qui sont associées au développement des commissures de la corde ventrale et qui jouent un rôle essentiel dans l’axogenèse [6]. Toujours chez D. melanogaster, Kuzbanian ou ADAM-10 (a disintegrin and metalloproteinase domain 10), une métalloprotéase appartenant à la famille des adamalysines1, interviendrait dans le guidage axonal et permettrait la répulsion des cônes de croissance en modulant, par clivage protéolytique, les interactions entre les éphrines et leurs récepteurs [7], des protéines impliquées dans l’axogenèse.
(→) m/s 2001, n° 6-7, p. 744 et 2003, n° 11, p. 1062
Le système MMP/TIMP dans différents processus liés aux maladies du système nerveux
L’expression des MMP et des TIMP dans le système nerveux de l’animal adulte sain est en général neuronale. Cependant, au cours de différentes maladies du système nerveux, la neuroglie réactive, les cellules de la barrière hématoencéphalique (BHE) et les cellules infiltrantes du système immunitaire deviennent une source importante de ces protéines. Cette modulation spatiotemporelle de l’expression du système MMP/TIMP et l’activité protéolytique nette qui en résulte ont été impliquées dans l’excitotoxicité, la mort neuronale, la perméabilisation de la BHE, la neuro-inflammation et la démyélinisation (Figure 1).
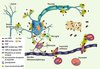 |
Figure 1. Rôles et mécanismes d’action potentiels des métalloprotéases matricielles (MMP) dans la physiopathologie du système nerveux central. 1. Rupture de la barrière hématoencéphalique (BHE) et infiltration de leucocytes : les MMP participent à la réponse inflammatoire dans le cerveau ainsi qu’aux interactions neurones/cellules gliales, en contrôlant l’activité des cytokines (membres de la famille du TNF, tumor necrosis factor, et interleukines), des molécules d’adhérence cellulaire (CAM) et des chimiokines (fractalkine). Les MMP fragilisent la BHE et facilitent l’infiltration de leucocytes (macrophages, lymphocytes T, polynucléaires neutrophiles…), qui participent à la réponse neuro-inflammatoire. 2. Déstabilisation de la matrice extracellulaire (MEC), des interactions avec le cytosquelette et des interactions cellule-cellule : une activité métalloprotéase non contrôlée déstabilise la matrice extracellulaire et altère les interactions cellule-matrice et protéines d’adhérence (CAM)-cytosquelette pour induire la mort neuronale. Inversement, l’inactivation par protéolyse de facteurs d’apoptose comme FasL (ligand de Fas) a des effets neuroprotecteurs. 3. Démyélinisation : les MMP dégradent la protéine basique de la myéline, substrat des MMP-1, -2, -3, -7 et -9. Cette activité métalloprotéase au niveau des oligodendrocytes contribuerait au processus pathologique dans la sclérose en plaques. 4. Plaques séniles : une activité réduite des MMP peut contribuer à des processus de fibrose provoqués par l’accumulation de substrats toxiques, comme le peptide β-amyloïde des plaques séniles de la maladie d’Alzheimer. L’accumulation du peptide serait facilitée par une augmentation des concentrations de TIMP-1 (tissue inhibitors of metalloproteinases-1) et une activation moindre de la MMP-9 (accumulée sous sa forme latente). 5. Croissance dendro-axonique, plasticité neuronale : les MMP participent à la croissance des neurites, à l’axogenèse, au guidage des cônes de croissance dans l’environnement matriciel, au cours du développement ou dans la plasticité réactive. |
Excitotoxicité et mort neuronale
L’hyperactivité neuronale induite par un agent convulsivant comme le kaïnate (un analogue du glutamate utilisé pour induire des convulsions et produire des modèles animaux d’épilepsie), ou par une libération massive de glutamate à la suite d’une ischémie cérébrale, provoque des lésions neuronales irréversibles de type excitotoxique. Ces lésions sont apparentes dans différentes structures cérébrales, notamment dans l’hippocampe dont les neurones pyramidaux (mais pas les neurones des grains) sont particulièrement vulnérables. Chez le rat, l’expression de TIMP-1 est fortement augmentée dans les neurones des grains de l’hippocampe quelques minutes après la reperfusion ischémique ou le déclenchement de crises convulsives. Quelques jours plus tard, la synthèse de TIMP-1 est induite dans des astrocytes réactifs proches des neurones lésés, mais pas dans la microglie réactive [8, 9]. Il a également été décrit une augmentation des concentrations de MMP-9 et -2 après une crise d’épilepsie ou une ischémie cérébrale chez le rat [9–11].
S’il reste important de poursuivre l’étude de l’expression spatio-temporelle des MMP et de leurs inhibiteurs dans le système nerveux, notamment au cours des processus pathologiques, seules les mesures d’activité protéolytique permettant d’évaluer le bilan net de la co-expression de protéases et d’inhibiteurs ont une signification sur le plan fonctionnel. Avec d’autres équipes, nous avons donc adapté les techniques de zymographie in situ pour étudier dans le système nerveux, et avec une résolution cellulaire, les modulations de l’activité des MMP au cours de différents processus pathologiques. Des données obtenues chez le rat par ces méthodes indiquent qu’après une ischémie cérébrale [9] ou des convulsions épileptiques, une augmentation de l’activité des MMP a lieu dans un premier temps dans les neurones, avant leur mort, et ultérieurement dans les cellules gliales réactives (Figure 2).
 |
Figure 2. Augmentation de l’activité métalloprotéase nette, visualisée par zymographie in situ après convulsions épileptiques et ischémie cérébrale. La technique de zymographie in situ consiste à appliquer sur des coupes fraîches de tissu cérébral de la gélatine (substrat des gélatinases endogènes) couplée à la fluorescéine piégée. Lorsque les MMP des cellules clivent ce substrat, la fluorescéine est libérée et visualisée au microscope. L’intensité du signal fluorescent augmente avec l’activité protéolytique nette, résultante de l’équilibre entre protéases et inhibiteurs endogènes. A. Activité gélatinolytique constitutive dans l’hippocampe de rat témoin. B. Une hyperactivité neuronale (crises épileptiques) provoque une augmentation de l’activité gélatinolytique nette dans l’hippocampe de rat, 6 heures après l’injection d’un agent convulsivant, le kaïnate. C. L’activité gélatinolytique est inhibée par un inhibiteur de métalloprotéases, la phénanthroline. D. Activité gélatinolytique constitutive dans l’aire CA1 de l’hippocampe d’un rat témoin non ischémié (chirurgie identique aux rats ischémiés). E. Trois jours après une ischémie globale transitoire, on observe une augmentation importante de l’activité gélatinolytique associée, d’une part, aux neurones pyramidaux (p) en cours de dégénérescence et, d’autre part, aux cellules gliales réactives facilement repérées à l’extérieur de la couche de neurones pyramidaux, dans le strata oriens (so) et radiatum (sr). On notera également une activité gélatinolytique importante dans les vaisseaux sanguins (flèches rouges), probablement en relation avec une fragilisation de la barrière hématoencéphalique. Échelle : A-C, 100 µm ; D, E, 200 µm (D et E, d’après [9]). |
Même si les mécanismes ne sont pas connus, les MMP peuvent contribuer de manière directe à la mort neuronale comme l’a démontrée l’application de MMP-9 sur des explants d’hippocampe [11] et de MMP-1 sur des explants de moelle épinière [12]. Dans les situations pathologiques telles que l’ischémie cérébrale, le monoxyde d’azote (NO) est augmenté dans le tissu nerveux ; la MMP-9 dont la forme latente est activée par le NO induit la mort des neurones corticaux, éventuellement par anoikis, une forme particulière d’apoptose qui résulte de la perte des interactions cellule-matrice [13]. Un mécanisme alternatif de mort neuronale induit par un stimulus excitotoxique met en jeu le clivage par les MMP de la fractalkine, une chimiokine exprimée principalement à la surface des neurones. La forme soluble de la fractalkine attire vers les neurones « atteints » des cellules microgliales réactives de type macrophages cytotoxiques [14] ; dans ce cas, l’action neurotoxique des MMP est indirecte et liée à leur rôle modulateur de la communication intercellulaire.
Inversement, les MMP peuvent aussi jouer un rôle neuroprotecteur lorsqu’elles modifient l’activité biologique de molécules qui engagent les cellules dans des voies apoptotiques. À titre d’exemple, les effets neurotoxiques du peptide β-amyloïde peuvent être inhibés ou potentialisés selon que le ligand de Fas (FasL), qui induit la mort cellulaire par apoptose après interaction avec Fas, est clivé ou non par des MMP, et notamment par MMP-7 [15]. Par ailleurs, il a été montré qu’en l’absence de son ligand, la nétrine-1 (voir ci-dessus), le récepteur DCC provoque l’apoptose [16]. Sachant que DCC est l’une des cibles des MMP, le clivage de ce récepteur pourrait protéger les cellules neurales en l’absence de nétrine-1.
Perméabilisation de la barrière hématoencéphalique, infiltration leucocytaire dans le système nerveux et neuro-inflammation
L’intégrité de la BHE, considérée comme la première ligne de défense du système nerveux, est essentielle. De nombreuses études impliquent les MMP, MMP-2 et -9 en particulier, dans la rupture de la BHE lors d’un processus neuropathologique. L’injection intracérébrale de MMP-2 fragilise la BHE alors que des anticorps bloquants de MMP-9 ou des inhibiteurs de MMP (IMMP) la protègent après une ischémie cérébrale (pour revue, voir [3]) ou un traumatisme de la moelle épinière [17]. La BHE des souris dont le gène de la MMP-9 a été invalidé est moins fragile après une ischémie cérébrale que celle des souris sauvages [18]. Dans ces situations pathologiques, les macrophages périvasculaires activés, qui expriment des concentrations élevées de MMP-9 et -2, participent certainement à la dégradation de la lame basale, riche en collagène IV. Les MMP exprimées par les leucocytes circulants facilitent leur extravasation à partir du sang ou de la lymphe. Ainsi, l’interaction de MMP-2 lymphocytaire avec l’intégrine a4 de la surface endothéliale est déterminante pour l’extravasation des lymphocytes T et l’infiltration du parenchyme nerveux dans l’encéphalomyélite auto-immune expérimentale (EAE), un modèle de sclérose en plaques (SEP) chez le rat [19].
Chez des patients atteints de SEP ou chez les rats soumis à une EAE, les concentrations de MMP-9 sont très élevées dans le liquide céphalo-rachidien. MMP-9, -1, -2 et -3 sont exprimées au niveau des plaques de démyélinisation chez les patients. Dans l’EAE, des concentrations élevées d’ARNm MMP-9 et -7 coïncident avec le pic de sévérité de la maladie. La modulation de l’expression de MMP et de TIMP dans le système nerveux pathologique est étroitement liée à la production et à la libération dans le parenchyme nerveux de facteurs trophiques et de cytokines pro-inflammatoires, qui sont des inducteurs (ou des répresseurs dans une moindre mesure) efficaces du système MMP/TIMP. Outre leur rôle dans la fragilisation de la BHE et la neuro-inflammation, certaines MMP - et notamment MMP-1, -2, -3, -7 et -9 - ont été impliquées dans la démyélinisation associée à la SEP ou à l’EAE puisqu’elles dégradent la protéine basique de la myéline (MBP). La digestion de la MBP par MMP-9 expose des épitopes immunodominants, dont l’injection suffit à induire l’EAE chez le rat (pour revue, voir [20]). La surexpression de MMP-3 chez une souris transgénique déclenche une maladie démyélinisante spontanée [21]. Dans l’EAE ou à la suite de lésions de la moelle épinière, les processus de démyélinisation sont atténués par des IMMP qui permettent respectivement une amélioration sensible des signes cliniques (pour revue, voir [3]) et de la locomotion [17].
Les troubles neurologiques causés par des infections virales sont accompagnés de processus neuro-inflammatoires avec là encore altération des équilibres au sein du système MMP/TIMP. Des niveaux élevés de MMP-9 ont été trouvés dans le liquide céphalo-rachidien des patients atteints de myélopathie progressive chronique (TSP/HAM) liée au virus humain lymphotrophique-1 (HTLV-I). In vitro, les lymphocytes T infiltrants infectés activent les astrocytes avec pour conséquences une augmentation de l’expression de MMP-3 et -9 [22]. Les causes des démences associées aux infections par le virus d’immunodéficience humaine-1 (VIH-1) n’ont pas été clairement déterminées, mais l’induction des MMP par différentes protéines virales et l’atténuation de leur toxicité par des IMMP suggèrent également une contribution du système MMP/TIMP aux processus neurodégénératifs liés au Sida (pour revue, voir [3, 23]).
Perspectives
L’idée simpliste selon laquelle les MMP seraient essentiellement impliquées dans la dégradation des protéines et donc délétères, alors que leurs inhibiteurs, endogènes ou de synthèse, auraient des effets plutôt bénéfiques laisse progressivement place à une vision plus complexe de la biologie du système MMP/TIMP. Il devient évident que les MMP modulent l’activité de nombreuses protéines extracellulaires et sont de véritables régulateurs de l’environnement cellulaire. Une meilleure connaissance de la régulation des MMP dans les différentes situations physiopathologiques et de leurs substrats matriciels, solubles ou membranaires, représente un véritable défi pour la recherche des prochaines années. Comme pour d’autres maladies, notamment celles à composante inflammatoire et dégénérative, les recherches entreprises dans les maladies du système nerveux indiquent que les MMP deviennent des cibles pharmacologiques importantes ; on pourrait donc assister au développement de nouvelles stratégies thérapeutiques fondées sur des inhibiteurs spécifiques, qui, de surcroît, passent la BHE.
Remerciements
Les travaux des auteurs sont subventionnés par le Cnrs, par l’Université de la Méditerranée Aix-Marseille II et par l’Association pour la Recherche sur la Sclérose en Plaques (ARSEP). Jérôme Jourquin et Crystel Ogier sont respectivement allocataires d’une bourse de thèse du Ministère de la Recherche et de la Ligue Nationale de Lutte Contre le Cancer.
Famille des protéines ADAM.
Références
- Vaillant C, Didier-Bazes M, Hutter A, Belin MF, Thomasset N. Spatiotemporal expression patterns of metalloproteinases and their inhibitors in the postnatal developing rat cerebellum. J Neurosci 1999; 19 : 4994–5004. [Google Scholar]
- Jaworski DM. Developmental regulation of membrane type-5 matrix metalloproteinase (MT5-MMP) expression in the rat nervous system. Brain Res 2000; 860 : 174–7. [Google Scholar]
- Rivera S, Khrestchatisky M. Matrix metalloproteinases and tissue inhibitors of metalloproteinases in neuronal plasticity and pathology. In : Baudry M, Thomsom RF, Davis JL, eds. Advances in synaptic plasticity. Cambridge, MA : The MIT Press, 1999 : 53–86. [Google Scholar]
- Galko MJ, Tessier-Lavigne M. Function of an axonal chemoattractant modulated by metalloprotease activity. Science 2000; 289 : 1365–7. [Google Scholar]
- Webber CA, Hocking JC, Yong VW, Stange CL, McFarlane S. Metalloproteases and guidance of retinal axons in the developing visual system. J Neurosci 2002; 22 : 8091–100. [Google Scholar]
- Llano E, Pendas AM, Aza-Blanc P, Kornberg TB, Lopez-Otin C. Dm1-MMP, a matrix metalloproteinase from Drosophila with a potential role in extracellular matrix remodeling during neural development. J Biol Chem 2000; 275 : 35978–85. [Google Scholar]
- Hattori M, Osterfield M, Flanagan JG. Regulated cleavage of a contact-mediated axon repellent. Science 2000; 289 : 1360–5. [Google Scholar]
- Rivera S, Tremblay E, Timsit S, Canals O, Ben-Ari Y, Khrestchatisky M. Tissue inhibitor of metalloproteinases-1 (TIMP-1) is differentially induced in neurons and astrocytes after seizures : evidence for developmental, immediate early gene, and lesion response. J Neurosci 1997; 17 : 4223–35. [Google Scholar]
- Rivera S, Ogier C, Jourquin J, et al. Gelatinase B and TIMP-1 are regulated in a cell- and time dependent manner in association with neuronal death and glial reactivity after global forebrain ischemia. Eur J Neurosci 2002; 15 : 19–32. [Google Scholar]
- Szklarczyk A, Lapinska J, Rylski M, McKay RD, Kaczmarek L. Matrix metalloproteinase-9 undergoes expression and activation during dendritic remodeling in adult hippocampus. J Neurosci 2002; 22 : 920–30. [Google Scholar]
- Jourquin J, Tremblay E, Decanis N, et al. Neuronal activity-dependent increase of net matrix metalloproteinase activity is associated with MMP-9 neurotoxicity after kainate. Eur J Neurosci 2003; 18 : 1507–17. [Google Scholar]
- Vos CM, Sjulson L, Nath A, et al. Cytotoxicity by matrix metalloprotease-1 in organotypic spinal cord and dissociated neuronal cultures. Exp Neurol 2000; 163 : 324–30. [Google Scholar]
- Gu Z, Kaul M, Yan B, et al. S-nitrosylation of matrix metalloproteinases : signaling pathway to neuronal cell death. Science 2002; 297 : 1186–90. [Google Scholar]
- Chapman GA, Moores K, Harrison D, Campbell CA, Stewart BR, Strijbos PJ. Fractalkine cleavage from neuronal membranes represents an acute event in the inflammatory response to excitotoxic brain damage. J Neurosci 2000; 20 : RC87. [Google Scholar]
- Ethell DW, Kinloch R, Green DR. Metalloproteinase shedding of Fas ligand regulates beta-amyloid neurotoxicity. Curr Biol 2002; 12 : 1595–600. [Google Scholar]
- Mehlen P, Rabizadeh S, Snipas SJ, Assa-Munt N, Salvesen GS, Bredesen DE. The DCC gene product induces apoptosis by a mechanism requiring receptor proteolysis. Nature 1998; 395 : 801–4. [Google Scholar]
- Noble LJ, Donovan F, Igarashi T, Goussev S, Werb Z. Matrix metalloproteinases limit functional recovery after spinal cord injury by modulation of early vascular events. J Neurosci 2002; 22 : 7526–35. [Google Scholar]
- Asahi M, Wang X, Mori T, et al. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J Neurosci 2001; 21 : 7724–32. [Google Scholar]
- Graesser D, Mahooti S, Madri JA. Distinct roles for matrix metalloproteinase-2 and alpha4 integrin in autoimmune T cell extravasation and residency in brain parenchyma during experimental autoimmune encephalomyelitis. J Neuroimmunol 2000; 109 : 121–31. [Google Scholar]
- Opdenakker G, Van den Steen PE, Van Damme J. Gelatinase B : a tuner and amplifier of immune functions. Trends Immunol 2001; 22 : 571–9. [Google Scholar]
- D’Souza CA, Mak B, Moscarello MA. The up-regulation of stromelysin-1 (MMP-3) in a spontaneously demyelinating transgenic mouse precedes onset of disease. J Biol Chem 2002; 277 : 13589–96. [Google Scholar]
- Giraudon P, Szymocha R, Buart S, et al. T lymphocytes activated by persistent viral infection differentially modify the expression of metalloproteinases and their endogenous inhibitors, TIMPs, in human astrocytes : relevance to HTLV-I-induced neurological disease. J Immunol 2000; 164 : 2718–27. [Google Scholar]
- Yong, VW, Power C, Forsyth P, Edwards DR. Metalloproteinases in biology and pathology of the nervous system. Nat Rev Neurosci 2001; 2 : 502–11. [Google Scholar]
Liste des figures
|
Figure 1. Rôles et mécanismes d’action potentiels des métalloprotéases matricielles (MMP) dans la physiopathologie du système nerveux central. 1. Rupture de la barrière hématoencéphalique (BHE) et infiltration de leucocytes : les MMP participent à la réponse inflammatoire dans le cerveau ainsi qu’aux interactions neurones/cellules gliales, en contrôlant l’activité des cytokines (membres de la famille du TNF, tumor necrosis factor, et interleukines), des molécules d’adhérence cellulaire (CAM) et des chimiokines (fractalkine). Les MMP fragilisent la BHE et facilitent l’infiltration de leucocytes (macrophages, lymphocytes T, polynucléaires neutrophiles…), qui participent à la réponse neuro-inflammatoire. 2. Déstabilisation de la matrice extracellulaire (MEC), des interactions avec le cytosquelette et des interactions cellule-cellule : une activité métalloprotéase non contrôlée déstabilise la matrice extracellulaire et altère les interactions cellule-matrice et protéines d’adhérence (CAM)-cytosquelette pour induire la mort neuronale. Inversement, l’inactivation par protéolyse de facteurs d’apoptose comme FasL (ligand de Fas) a des effets neuroprotecteurs. 3. Démyélinisation : les MMP dégradent la protéine basique de la myéline, substrat des MMP-1, -2, -3, -7 et -9. Cette activité métalloprotéase au niveau des oligodendrocytes contribuerait au processus pathologique dans la sclérose en plaques. 4. Plaques séniles : une activité réduite des MMP peut contribuer à des processus de fibrose provoqués par l’accumulation de substrats toxiques, comme le peptide β-amyloïde des plaques séniles de la maladie d’Alzheimer. L’accumulation du peptide serait facilitée par une augmentation des concentrations de TIMP-1 (tissue inhibitors of metalloproteinases-1) et une activation moindre de la MMP-9 (accumulée sous sa forme latente). 5. Croissance dendro-axonique, plasticité neuronale : les MMP participent à la croissance des neurites, à l’axogenèse, au guidage des cônes de croissance dans l’environnement matriciel, au cours du développement ou dans la plasticité réactive. |
| Dans le texte | |
|
Figure 2. Augmentation de l’activité métalloprotéase nette, visualisée par zymographie in situ après convulsions épileptiques et ischémie cérébrale. La technique de zymographie in situ consiste à appliquer sur des coupes fraîches de tissu cérébral de la gélatine (substrat des gélatinases endogènes) couplée à la fluorescéine piégée. Lorsque les MMP des cellules clivent ce substrat, la fluorescéine est libérée et visualisée au microscope. L’intensité du signal fluorescent augmente avec l’activité protéolytique nette, résultante de l’équilibre entre protéases et inhibiteurs endogènes. A. Activité gélatinolytique constitutive dans l’hippocampe de rat témoin. B. Une hyperactivité neuronale (crises épileptiques) provoque une augmentation de l’activité gélatinolytique nette dans l’hippocampe de rat, 6 heures après l’injection d’un agent convulsivant, le kaïnate. C. L’activité gélatinolytique est inhibée par un inhibiteur de métalloprotéases, la phénanthroline. D. Activité gélatinolytique constitutive dans l’aire CA1 de l’hippocampe d’un rat témoin non ischémié (chirurgie identique aux rats ischémiés). E. Trois jours après une ischémie globale transitoire, on observe une augmentation importante de l’activité gélatinolytique associée, d’une part, aux neurones pyramidaux (p) en cours de dégénérescence et, d’autre part, aux cellules gliales réactives facilement repérées à l’extérieur de la couche de neurones pyramidaux, dans le strata oriens (so) et radiatum (sr). On notera également une activité gélatinolytique importante dans les vaisseaux sanguins (flèches rouges), probablement en relation avec une fragilisation de la barrière hématoencéphalique. Échelle : A-C, 100 µm ; D, E, 200 µm (D et E, d’après [9]). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.