")
")
| Issue |
Med Sci (Paris)
Volume 19, Number 12, Décembre 2003
|
|
|---|---|---|
| Page(s) | 1226 - 1232 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/200319121226 | |
| Published online | 15 December 2003 | |
Effets vasculaires des œstrogènes
Usefulness of experimental models to understand the vascular effects of estrogens
1
Inserm U. 589 et Physiologie Médicale, CHU Rangueil, TSA 50032, 31059 Toulouse Cedex, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
Le traitement hormonal substitutif ne confère pas de protection cardio-vasculaire chez la femme ménopausée comme le montre l’étude nord-américaine WHI 2002 (Women’s health initiative). Ce résultat contraste avec le fait que l’œstradiol (E2) prévient la constitution de la strie lipidique (c’est-à-dire des premiers stades de l’athérosclérose) dans tous les modèles animaux. L’effet bénéfique sur le profil lipidique, observé en clinique, est absent chez la souris et ne peut donc expliquer l’effet protecteur observé dans cette espèce. Les œstrogènes exercent un effet bénéfique sur l’endothélium, comme l’augmentation de la production de monoxyde d’azote, l’accélération de la réendothélialisation ou encore la prévention de l’apoptose. Les effets de l’œstradiol sur le système immuno-inflammatoire sont plus ambigus. D’une part, alors que l’E2 protège des souris immunocompétentes du dépôt lipidique, il n’a plus d’effet chez des souris immunodéficientes. Ainsi, les lymphocytes sont nécessaires à la manifestation des effets bénéfiques de l’E2 aux premiers stades du processus athéromateux (constitution de la strie lipidique) dans tous les modèles expérimentaux. D’autre part, en augmentant la production d’interféron g, l’E2 pourrait contribuer à déstabiliser la chape fibro-musculaire qui encercle la strie lipidique. Cet effet potentiellement néfaste sur la stabilité de la plaque d’athérosclérose pourrait rendre compte de l’absence de bénéfice (et même d’un effet délétère à l’instauration du traitement) sur les lésions d’athérosclérose des femmes ménopausées.
Abstract
Hormonal replacement therapy does not prevent cardiovacular events in postmenopausal women. In contrast, the incidence of cardiovascular diseases is higher in men than in premenopausal women but increases in postmenopausal women, and all animal studies demonstrate a prevention of fatty streak deposit by estradiol. Although estradiol improves the lipoprotein profile, this effect can account for only a minor part of the protective effect. Endothelium appears to be an important target for estradiol, because this hormone potentiates endothelial nitric oxide (NO) production, thus promoting the beneficial effects of NO, such as vasorelaxation and inhibition of platelet aggregation. Estradiol accelerates endothelial regrowth, thus favoring vascular healing, and prevents apoptosis of endothelial cells. Estradiol prevents fatty streak deposit through a mechanism which is clearly independent of NO. The immuno-inflammatory system appears to play a key role in the development of fatty streak deposit as well as in atherosclerotic plaque rupture. Mice deficient either in monocyte-macrophages or in lymphocytes are partially protected against fatty streak deposit. Interestingly, the atheroprotective effect of estradiol is absent in mice deficient in T and B lymphocytes. Most of these effects of estradiol are mediated by estrogen receptor a, and are independent of estrogen receptor b. Thus, the inflammatory-immune system appears to be also a major target of estrogens. However, the effects of estrogens on the immuno-inflammatory system appear ambiguous, as in some models, estradiol rather promotes inflammation (by increasing interferon g which could elicit plaque destabilization). A better understanding of the mechanisms of estrogens on the normal and atheromatous arteries is required and should help to optimize the prevention of cardiovascular disease after menopause.
© 2003 médecine/sciences - Inserm / SRMS
Les résultats de l’étude nord-américaine WHI (women’s health initiative) [1] visant à évaluer les bénéfices apportés par un traitement hormonal substitutif chez les femmes ménopausées sans antécédent cardio-vasculaire ont été publiés au cours de l’été 2002. Ils confirment l’accroissement modeste mais significatif du risque de cancer du sein sous traitement hormonal substitutif. Cette étude révèle qu’un traitement hormonal substitutif ne confère pas de protection cardio-vasculaire, et même accroît légèrement le risque d’accident cardio-vasculaire dans l’année qui suit l’instauration du traitement hormonal (‹).
(‹) m/s 2002, n° 11, p. 1049
Ces résultats sont très superposables à ceux de l’étude HERS (heart and estrogen/progestin replacement study) parue en 1998, évaluant le traitement hormonal substitutif en prévention secondaire chez des femmes ménopausées ayant un antécédent d’accident cardiovasculaire [2]. Il ne faut cependant pas perdre de vue que le premier but d’un traitement hormonal substitutif est de supprimer les troubles fonctionnels comme les troubles vasomoteurs (bouffées de chaleur, sueurs nocturnes) ou les troubles trophiques génito-urinaires qui apparaissent à la ménopause, le deuxième bénéfice étant la prévention de l’ostéoporose.
Ces deux grandes études d’intervention ont été suscitées par des études d’observation montrant que le risque cardiovasculaire chez des femmes entre 35 et 65 ans est significativement plus bas que celui des hommes de même âge, et que le risque des femmes rejoint progressivement celui des hommes après la ménopause. Cela suggérait que l’œstradiol endogène (E2) pouvait être responsable, au moins en partie, de cet effet athéroprotecteur. Cette hypothèse est confortée par l’effet protecteur de l’œstradiol dans tous les modèles animaux d’athérosclérose [3]. Ainsi, chez des animaux soumis à un régime hypercholestérolémique [3] comme chez les souris rendues hypercholestérolémiques par inactivation génique [4], l’œstradiol prévient la constitution de la strie lipidique, c’est-à-dire des premiers stades de l’athérosclérose.
Ainsi, il semble exister une discordance entre l’effet des œstrogènes chez la femme ménopausée et l’effet des œstrogènes chez la femme en période d’activité génitale ainsi que dans tous les modèles expérimentaux. Dans cette revue, nous résumerons dans un premier temps la compréhension que nous avons actuellement de l’effet des œstrogènes sur la paroi vasculaire normale et pathologique. Cela nous amènera à discuter dans un deuxième temps ce que l’on peut attendre de l’effet des œstrogènes sur la paroi artérielle et quelles explications on peut actuellement proposer concernant la divergence des effets dans les modèles expérimentaux et en clinique.
Modèles d’athérosclérose et œstradiol
L’athérosclérose est une maladie touchant les artères de grand et moyen calibre caractérisée par l’accumulation de lipides à la faveur d’un processus inflammatoire chronique [5] (‹) (Figure 1).
(‹) m/s 2001, n° 2, p. 162
 |
Figure 1. Les différentes étapes de la constitution de la strie lipidique et de la plaque d’athérosclérose. La première étape est la pénétration de lipoprotéines athérogènes, en particulier des lipoprotéines de faible densité (LDL) à travers la monocouche de cellules endothéliales. L’oxydation des LDL dans l’espace sous-endothélial représente probablement une modification nécessaire aux étapes ultérieures, car les LDL oxydées induisent à leur tour une activation de l’endothélium, en particulier l’expression de molécules d’adhérence pour les leucocytes. Ces molécules d’adhérence endothéliales sont nécessaires pour ralentir les monocytes circulants, les arrêter et permettre leur migration dans l’intima. Les monocytes sont alors activés en macrophages (Ma) ce qui contribue probablement à accroître l’oxydation des LDL (flèches pointillées). Les LDL ainsi davantage oxydées (oxLDL) peuvent être reconnues par des récepteurs éboueurs ( ) (scavenger) présents sur ces macrophages. En fonction de nombreux facteurs systémiques comme les facteurs de risques classiques (hypertension artérielle, hypercholestérolémie, tabagisme, diabète) et de facteurs locaux (degré de dysfonctionnement endothélial, chimiokines, cytokines produites par les différents acteurs cellulaires [endothélium, monocytes-macrophages, cellules musculaires lisses mais aussi lymphocytes]), les macrophages nettoient l’intima et préviennent ainsi l’accumulation de LDL oxydées, ou s’accumulent et deviennent progressivement spumeux (Mas), constituant progressivement la strie lipidique. L’athérome est ainsi à la fois une pathologie métabolique et inflammatoire. L’extension de la strie lipidique tend à être limitée par une réaction cicatricielle des cellules musculaires lisses (CML) de la média qui migrent dans l’intima et sécrètent du collagène (Co). Le niveau d’inflammation de la strie lipidique et la solidité de la chape fibro-musculaire conditionnent la stabilité de la plaque d’athérosclérose, dont la rupture expose des matériaux thrombogènes à l’origine de la formation d’un thrombus, qui menace le territoire artériel en aval en cas d’occlusion de la lumière vasculaire. |
Ainsi, l’hypercholestérolémie conduit à la rétention prolongée de lipoprotéines de basse densité dans l’espace sous-endothélial qui vont s’agréger, s’oxyder, activer l’endothélium sus jacent et provoquer une réponse athérogène au niveau de sites de prédilection à la faveur d’anomalies de contrainte hémodynamique (‹).
(‹) m/s 2001, n° 5, p. 559
Ces anomalies sont le plus souvent représentées par un flux sanguin turbulent qui se produit au niveau des bifurcations artérielles. Comme nous l’avons évoqué précédemment, un traitement chronique par l’œstradiol prévient de façon constante la strie lipidique dans tous les modèles animaux d’athérosclérose. Ces effets ont été récemment l’objet d’une revue de la littérature [3]. Chez des lapins et des singes soumis à un régime riche en cholestérol, le traitement par l’œstradiol de femelles mais aussi de mâles castrés entraîne une protection que traduit la réduction de 35 à 80 % de la taille des lésions au niveau de l’aorte et des artères coronaires [3]. L’ajout d’un progestatif n’a pas d’effet dans certaines études, mais peut atténuer l’effet des œstrogènes dans d’autres études.
Avec l’avènement de l’inactivation génique par recombinaison homologue, la souris est devenue un modèle clé pour identifier les gènes et les voies impliqués dans l’athérogenèse. Des souris rendues déficientes soit en apolipoprotéine E, soit en récepteur des LDL, développent spontanément des lésions athéromateuses qui, avec le temps, deviennent complexes et ressemblent beaucoup aux lésions d’athérosclérose humaine. Dans ces deux modèles, les œstrogènes inhibent la constitution de la strie lipidique de façon marquée (en moyenne 75 % de réduction des lésions) [4]. L’œstradiol endogène confère un certain degré de protection comme en témoigne l’effet aggravant de la castration sur le développement de la strie lipidique. Cependant, les études de relation dose/réponse ont permis de montrer que des doses d’œstradiol du niveau de celles rencontrées au cours de la gestation sont nécessaires pour obtenir un effet protecteur maximal [6]. Le traitement par l’œstradiol s’associe à une réduction de la cholestérolémie totale dans un certain nombre d’études (mais pas dans toutes [4]). En fait, cette baisse de la cholestérolémie est surtout due à une baisse du cholestérol contenu dans les lipoprotéines de haute densité (ou HDL cholestérol, qui est protecteur) et ne peut donc expliquer l’effet anti-athéromateux observé dans cette espèce [6].
Il faut aussi rappeler la limite actuelle des modèles expérimentaux dans la problématique de l’athérosclérose. Les premiers stades de l’athérosclérose sont totalement asymptomatiques. C’est seulement la rupture de plaque ou l’érosion de l’endothélium suivie d’une thrombose obstructive de l’artère, qui, en provoquant une ischémie du territoire d’aval, font toute la dangerosité de cette maladie. Les essais cliniques comme les études HERS ou WHI enregistrent donc ces événements tout à fait terminaux du processus athéroscléreux. Malheureusement, la rupture de plaque suivie de thrombose est un événement trop rare dans les modèles de souris athéromateuses pour pouvoir être étudié. Ainsi nous n’avons pas de preuve qu’un traitement qui par exemple est bénéfique sur les premiers stades de l’athérosclérose le soit aussi à des stades plus tardifs comme la rupture de plaque. Nous y reviendrons.
Œstradiol, récepteurs des œstrogènes et athérosclérose
Le spasme artériel et la thrombose jouent un rôle majeur dans le déclenchement des accidents cardio-vasculaires. L’endothélium produit de nombreuses substances, dont le monoxyde d’azote (NO), un messager radicalaire qui joue un rôle vasculoprotecteur important du fait de ses propriétés vaso-dilatatrices et anti-agrégantes. Les travaux de Gisclard et al. ont les premiers mis en évidence une potentialisation par l’œstradiol de la production endothéliale de NO au niveau de l’artère fémorale de lapin. Dans cette espèce, l’œstradiol potentialise la relaxation endothélium dépendante en réponse à l’acétylcholine. Ainsi, une augmentation de la production basale et/ou stimulée de NO a été observée dans un certain nombre de territoires (mais pas dans tous) chez la plupart des espèces, et il a été initialement proposé que cet effet contribue de façon majeure à l’effet athéroprotecteur de l’œstradiol. Nous avons pour notre part étudié l’effet de l’œstradiol sur la production endothéliale de NO chez la souris. Nous avons pu montrer que, comme dans d’autres espèces, l’œstradiol augmente la production basale de NO par l’endothélium.
Les effets de l’œstradiol requièrent probablement des changements de l’expression génique (effets génomiques) bien que des effets à court terme indépendants de la transcription (effets extragénomiques) aient été aussi rapportés [7]. Le récepteur des œstrogènes a (ERa) est le récepteur « classique » des œstrogènes, qui est le médiateur essentiel des effets de ces hormones au cours de la reproduction [8]. Plus récemment, un deuxième récepteur appelé ERb a été identifié, mais son rôle demeure à l’heure actuelle mal connu. Il est surexprimé dans la carotide de rat après agression endovasculaire [9]. Nous avons comparé l’effet de l’œstradiol dans deux lignées de souris transgéniques invalidées pour ERa : une première lignée (ERa-Néo KO) [10] dans laquelle une cassette contenant l’ADNc conférant la résistance à la néomycine a été insérée dans le premier exon du gène ERa et une deuxième lignée (ERa-D2 KO) [11], consistant en l’excision du deuxième exon du gène ERa, codant pour le domaine de liaison du récepteur à l’ADN (Figure 2A). Comme attendu, la construction de la lignée ERa-D2 KO ne permet l’expression d’aucune protéine ERa. En revanche, nous avons mis en évidence chez les souris ERa-Néo KO, à la faveur de mécanismes d’épissages alternatifs déjà décrits [12], l’existence de deux formes tronquées de récepteurs : une isoforme de 46 kDa et une molécule chimère de 55 kDa contenant 7 acides aminés de la cassette néomycine (Figure 2B). Ces deux récepteurs sont dépourvus de la fonction transactrivatrice AF1, mais conservent la fonction transactrivatrice AF2 [13]. Parallèlement, nous avons étudié l’effet de l’œstradiol sur la production endothéliale de NO dans ces deux lignées de souris. Tandis que l’effet de l’œstradiol sur cette production est totalement aboli chez les souris ERa-D2 KO, l’effet de l’œstradiol persiste chez les souris ERa-Néo KO et est identique à celui observé chez les souris sauvages [14]. L’ensemble de ces résultats suggère que la fonction AF1 du ERa n’est probablement pas nécessaire à l’effet de l’œstradiol dans l’endothélium [14]. Au total, l’effet de l’œstradiol sur la production endothéliale de NO, mais aussi sur la réendothélialisation, dépend du récepteur des œstrogènes a (ERa), puisque ces deux effets sont abolis chez les souris rendues déficientes en ERa, alors qu’ils persistent chez les souris déficientes en ERb [15].
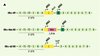  |
Figure 2. Constructions utilisées dans deux lignées transgéniques pour invalider le gène du récepteur des œstrogènes (ERa). A. Le gène ERa (ERa-WT) comporte 8 exons. Deux codons initiateurs ATG1 (dans l’exon 1) et ATG2 (dans l’exon 2) peuvent être utilisés, à l’origine d’isoformes de longueurs différentes. La partie codante du gène est précédée par une région 5’ non traduite (5’UTR). La première construction d’inactivation de ERa (ERa-Néo KO [10]) a consisté à insérer une cassette néomycine dans le premier exon du gène ERa. La deuxième construction d’inactivation de ERa (ERa-D2 KO [11]) a consisté à exciser le deuxième exon du gène ERa codant pour le domaine de liaison du récepteur à l’ADN. B. Le récepteur des œstrogènes a, ERa (66) comporte 6 domaines (de A à F), dont des domaines de liaison à l’ADN, à l’hormone œstrogène et des fonctions transactivatrices AF1 et AF2. La construction de la lignée ERa-D2 KO ne permet l’expression d’aucune protéine fonctionnelle en l’absence de domaine de liaison à l’ADN. En revanche, nous avons mis en évidence, chez les souris ERa-Néo KO, à la faveur de mécanismes d’épissage alternatif, l’existence de deux formes tronquées de récepteurs : une isoforme de 46 kDa (46) et une molécule chimère de 55 kDa (55) contenant 7 acides aminés de la cassette néomycine. Ces deux récepteurs sont certes dépourvus de la fonction transactivatrice AF1, mais conservent la fonction transactivatrice AF2. |
Étant donné que le blocage de la production de NO accélère la progression des stries lipidiques chez le lapin hypercholestérolémique, nous avons testé l’hypothèse selon laquelle l’augmentation de la production basale de NO par l’endothélium en réponse à l’œstradiol pourrait contribuer à son effet athéroprotecteur. Nous n’avons pas observé d’effet du blocage de la production de NO sur le développement de la strie lipidique chez la souris déficiente en apoE, à la différence de ce qui avait été observé chez le lapin [16]. De plus, l’effet protecteur de l’œstradiol n’est en rien altéré par le blocage de la production de NO, démontrant que l’augmentation de la production de NO n’est pas impliquée dans la prévention de la strie lipidique [16]. Néanmoins, cet effet est probablement bénéfique à des stades plus avancés de l’athérosclérose, car le NO est antispastique et antiagrégant plaquettaire, et protège donc des complications tardives de l’athérosclérose.
Les effets de l’œstradiol au niveau de l’endothélium ne se limitent pas à potentialiser la production de NO. Nous avons pu montrer récemment que l’œstradiol accélère la vitesse de réendothélialisation dans un modèle d’agression électrique de la carotide chez la souris [17]. On peut ainsi considérer que l’œstradiol favorise le processus de cicatrisation artérielle après une agression. L’œstradiol exerce d’autres effets bénéfiques sur l’endothélium en particulier. Il prévient ainsi l’apoptose induite par le TNFa [18]. Il a aussi été proposé que l’œstradiol puisse prévenir l’induction de molécules d’adhérence comme ICAM 1 (intercellular adhesion molecule 1) dans des cellules endothéliales en culture. Cependant, l’œstradiol prévient le dépôt lipidique chez des souris déficientes à la fois en apolipoprotéine E (apoE) et en molécules d’adhérence des leucocytes (soit ICAM1, soit P-sélectine) avec la même intensité qu’il le fait chez les souris déficientes en apoE [19]. Nous pouvons donc conclure que l’effet athéroprotecteur de l’œstradiol n’est pas en rapport avec un effet sur l’expression endothéliale d’une de ces deux molécules d’adhérence.
Comme nous l’avons vu précédemment, deux types d’invalidation de ERa (ERa-Néo KO et ERa-D2 KO) ont été réalisés et diffèrent de façon substantielle. Deux invalidations pour ERb ont aussi été réalisées, mais les souris mutantes apparaissent phénotypiquement identiques. Afin de déterminer la contribution relative de chacun de ces deux récepteurs dans l’effet athéroprotecteur de l’œstradiol, J.B. Hodgin et al. ont croisé les souris knock-out pour ERa et ERb avec des souris déficientes en apoE. L’œstradiol diminue les lésions d’athérosclérose des animaux apoE−/− de plus de 80 % tandis que l’effet inhibiteur de l’œstradiol est en grande partie aboli chez les souris double mutantes ERa−/−/ApoE−/− [20]. En revanche, l’œstradiol inhibe la progression et les lésions d’athérosclérose chez les souris ERb−/−/apoE−/− de la même façon que chez les souris apoE−/−, démontrant que l’effet athéroprotecteur de l’œstradiol est totalement conservé en l’absence d’ERb [21]. De plus, l’effet athéroprotecteur du soja alimentaire, une source de phyto-œstrogènes, s’exerce également par l’intermédiaire d’ERa puisque son effet est aboli chez les souris Era−/−/ApoE−/−. De façon intéressante, l’œstradiol diminue la formation de la chape fibromusculaire chez les souris apoE−/− mais aussi chez les souris Era−/−/ApoE−/−. Cette observation suggère la possibilité que ERb puisse jouer un rôle protecteur dans la maturation des plaques tandis qu’il n’est clairement pas impliqué dans des stades plus précoces comme le développement de la strie lipidique. Alternativement, cet effet résiduel de l’œstradiol pourrait être dû à ERa 55 chimérique dont l’expression persiste chez les souris Era−/− dont l’invalidation est imparfaite [14]. Cet effet de prévention de la formation de la chape fibromusculaire par l’œstradiol ne peut pas être considéré comme un effet bénéfique, mais pourrait au contraire être délétère, en empêchant le processus de cicatrisation de la strie lipidique et donc favoriser l’instabilité des plaques. Cet effet pourrait contribuer, s’il se produit chez la femme, à l’augmentation de la fréquence des accidents cardio-vasculaires dans les mois qui suivent l’instauration du traitement hormonal substitutif comme cela a pu être observé aussi bien dans l’étude HERS [2] que dans l’étude WHI [1].
Œstradiol et inflammation au cours de l’athérosclérose
Le captage des LDL (low density lipoproteins) oxydées par l’intermédiaire des récepteurs éboueurs des macrophages et la transformation ultérieure des macrophages en cellules spumeuses constituent un mécanisme essentiel de l’initiation et de la progression de la plaque athéromateuse (‹). Le système immunitaire favorise le processus athéromateux puisque des souris immunodéficientes (par inactivation de la recombinase RAG, ce qui provoque un déficit en lymphocytes B et T matures) développent deux fois moins de lésions que des souris immunocompétentes [22]. Nous avons exploré l’implication du système immunitaire dans l’effet athéroprotecteur de l’œstradiol. Nous avons montré que le système immunitaire est nécessaire à la manifestation de l’effet athéroprotecteur de l’œstradiol puisque, alors que ce dernier protège des souris immunocompétentes du dépôt lipidique, il ne prévient plus le développement des lésions chez des souris immuno-déficientes et hypercholestérolémiques (RAG-2−/− apoE−/−) [23]. Nous avons pu récemment confirmer ce caractère indispensable des lymphocytes pour l’effet athéroprotecteur dans un autre modèle d’athérosclérose (RAG-2−/− LDL récepteur−/−). Cette observation repose la question de la cible cellulaire de l’œstradiol à savoir l’endothélium ou le système immunitaire. En effet, les deux populations cellulaires pourraient être les médiateurs de l’effet de l’œstradiol puisque le passage trans-endothélial des cellules du système immuno-inflammatoire représente une étape clé du développement du processus athéromateux. Une partie de l’effet athéroprotecteur pourrait être due à un effet immunomodulateur de doses endogènes d’œstradiol, puisque l’on sait par exemple que ce niveau d’imprégnation hormonale suffit à diminuer le poids du thymus. Par ailleurs, tous les effets de l’œstradiol sur l’endothélium recensés à ce jour vont dans le sens d’effets bénéfiques. Cependant, ces effets semblent requérir des concentrations circulantes d’œstradiol du niveau de celles qui sont observées durant la gestation, tandis qu’ils n’apparaissent que modestement à des concentrations du niveau de celles du cycle œstral [6, 11, 15]. Cette hypothèse d’un recrutement d’un effet immunitaire pour de faibles doses d’œstradiol et d’un effet sur l’endothélium pour des doses plus élevées méritera d’être testée à l’aide de modèles appropriés (modèle Cre-Lox permettant l’inactivation spécifique du gène ERa dans l’endothélium [24] et modèle de greffe de moelle osseuse de souris déficientes en ERa).
Les mécanismes inflammatoires qui contribuent au processus athéromateux font, comme dans tout processus inflammatoire, intervenir des cytokines. Ces cytokines ont été classifiées, de façon un peu schématique, en cytokines pro- et anti-inflammatoires. On peut alors s’attendre à ce que le blocage de certaines cytokines pro-inflammatoires et/ou l’administration de certaines cytokines anti-inflammatoires aient un effet athéroprotecteur, et inversement (‹).
(‹) m/s 2001, n° 2, p. 162
Cela se vérifie par exemple pour l’interféron g ou l’interleukine-1 qui accélèrent le processus athéromateux [25], tandis que l’interleukine 10 s’avère protectrice [26]. Étant donné l’importance du système immunitaire dans l’effet athéroprotecteur de l’E2, l’implication voire la régulation d’une ou plusieurs cytokines par l’œstradiol représente donc une hypothèse qu’il nous a semblé logique de tester. Les cytokines candidates sont nombreuses, et leur choix a été orienté par les données de la littérature. Ainsi, l’interleukine-6 joue un rôle clé dans la déminéralisation osseuse secondaire à la castration. Néanmoins, l’interleukine-6 n’est pas impliquée dans l’effet athéroprotecteur de l’œstradiol, car des souris hypercholestérolémiques et déficientes en interleukine-6 sont protégées par l’œstradiol comme le sont des souris seulement hypercholestérolémiques [27]. Dans deux modèles permettant d’explorer spécifiquement le système immuno-inflammatoire, nous avons observé que l’œstradiol augmente la production d’interféron g respectivement par les lymphocytes T des ganglions drainant le site d’immunisation [28] et les lymphocytes NKT après activation spécifique de cette population. Nous testerons prochainement le rôle de l’interféron g dans l’effet de l’œstradiol sur la constitution de la strie lipidique. En outre, la production d’interféron g par les lymphocytes, mais aussi dans une moindre mesure par les macrophages, est susceptible de jouer un rôle important dans la stabilité des plaques [5, 28]. Si sous l’effet de l’œstradiol la production d’interféron g augmente au niveau de la plaque d’athérome, ce mécanisme pourrait contribuer à déstabiliser la chape fibromusculaire et à favoriser la rupture de plaque [29], rendant compte de l’augmentation de la fréquence des accidents cardiovasculaires dans les mois qui suivent l’instauration de ce traitement dans les deux études d’intervention HERS et WHI.
Conclusions et perspectives
Tandis que dans les modèles expérimentaux, l’œstradiol protège des premiers stades de l’athérome, les études d’intervention par traitement hormonal substitutif chez la femme ménopausée n’ont pas mis en évidence de protection, mais au contraire montrent même une aggravation du risque cardio-vasculaire dans la première année qui suit l’instauration du traitement. Cependant, les œstrogènes utilisés dans ces études nord-américaines (œstrogènes équins conjugués) et associés avec un progestatif (acétate de médroxyprogestérone) sont différents des hormones naturelles 17b œstradiol et progestérone utilisées en Europe ainsi que dans les modèles expérimentaux. De plus, la plupart de ces essais cliniques s’adressent à des femmes d’âge mûr dont certaines ont probablement des lésions athéroscléreuses avancées. Dans ce contexte, il est important de rappeler que trois des quatre études réalisées chez des animaux ayant des lésions préexistantes démontraient un effet atténué ou pas d’effet du traitement par l’œstradiol, suggérant que l’effet athéroprotecteur de l’œstradiol puisse être perdu une fois que les lésions d’athérosclérose sont établies [4].
Ainsi, dans l’état actuel de nos connaissances, les œstrogènes apparaissent bénéfiques sur les stades précoces de la constitution de la strie lipidique (modèles expérimentaux et potentiellement chez la femme en période d’activité génitale). Les cibles cellulaires de l’oestradiol impliquent probablement à la fois l’endothélium et le système immuno-inflammatoire, et l’élucidation des mécanismes cellulaires et moléculaires de l’effet athéroprotecteur de l’œstradiol pourrait permettre de mettre à jour de nouvelles cibles thérapeutiques.
En revanche, les œstrogènes apparaissent inefficaces à des stades tardifs du processus athéroscléreux, et des arguments indirects suggèrent qu’ils pourraient favoriser la rupture de la plaque athéromateuse (dont un mécanisme pourrait être l’augmentation de la production d’interféron g). Les études HERS et WHI soulignent la nécessité de comprendre les effets des œstrogènes à un niveau cellulaire et moléculaire. Cette démarche est d’autant plus indispensable que, dans l’état actuel de nos ressources thérapeutiques, seuls les œstrogènes permettent le traitement des troubles fonctionnels de la ménopause qui constituent la première indication du traitement hormonal substitutif.
Références
- Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA 2002; 288 : 321–33. [Google Scholar]
- Hulley S, Grady D, Bush T, et al. Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. Heart and estrogen/ progestin replacement study (HERS) research group. JAMA 1998; 280 : 605–13. [Google Scholar]
- Holm P. Effect of estrogen on development of atherosclerosis. A review of experimental animal studies. Dan Med Bull 2002; 48 : 146–60. [Google Scholar]
- Hodgin J, Maeda N. Estrogen and mouse models of atherosclerosis. Endocrinology 2002; 143 : 4495–501. [Google Scholar]
- Ross R. Atherosclerosis: an inflammatory disease. N Engl J Med 1999; 340 : 115–25. [Google Scholar]
- Elhage R, Arnal JF, Pierragi MT, et al. Estradiol-17b prevents fatty streak formation in apolipoprotein E-deficient mice. Arterioscl Thromb Vasc Biol 1997; 17 : 2679–84. [Google Scholar]
- Mendelsohn ME. Nongenomic, ER-mediated activation of endothelial nitric oxide synthase: how does it work ? What does it mean ? Circ Res 2000; 87 : 956–60. [Google Scholar]
- Couse JF, Korach KS. Estrogen receptor null mice : what have we learned and where will they lead us ? Endocrinol Rev 1999; 20 : 358–417. [Google Scholar]
- Makela S, Savolainen H, Aavik E, et al. Differentiation between vasculoprotective and uterotrophic effects of ligands with different binding affinities to estrogen receptors alpha and beta. Proc Natl Acad Sci USA 1999; 96 : 7077–82. [Google Scholar]
- Lubahn DB, Moyer JS, Golding TS, Couse JF, Korach KS, Smithies O. Alteration of reproductive function but not prenatal sexual development after insertional disruption of the mouse estrogen receptor gene. Proc Natl Acad Sci USA 1993; 90 : 11162–6. [Google Scholar]
- Dupont S, Krust A, Gansmuller A, Dierich A, Chambon P, Mark M. Effect of single and compound knockouts of estrogen receptors alpha (ERalpha) and beta (ERbeta) on mouse reproductive phenotypes. Development 2000; 127 : 4277–91. [Google Scholar]
- Couse JF, Curtis SW, Washburn TF, et al. Analysis of transcription and estrogen insensitivity in the female mouse after targeted disruption of the estrogen receptor gene. Mol Endocrinol 1995; 9 : 1441–54. [Google Scholar]
- Flouriot G, Brand H, Denger S, et al. Identification of a new isoform of the human estrogen receptor-alpha (hER-alpha) that is encoded by distinct transcripts and that is able to repress hER-alpha activation function 1. Embo J 2000; 19 : 4688–700. [Google Scholar]
- Pendaries C, Darblade B, Rochaix P, et al. The AF-1 activation-function of ERalpha may be dispensable to mediate the effect of estradiol on endothelial NO production in mice. Proc Natl Acad Sci USA 2002; 99 : 2205–10. [Google Scholar]
- Darblade B, Pendaries C, Krust A, et al. Estradiol alters nitric oxide production in the mouse aorta through the alpha-, but not beta-, estrogen receptor. Circ Res 2002; 90 : 413–9. [Google Scholar]
- Elhage R, Bayard F, Richard V, et al. The prevention of fatty streak formation of 17b-estradiol is not mediated by the production of nitric oxide in apolipoprotein E-deficient mice. Circulation 1997; 96 : 3048–52. [Google Scholar]
- Brouchet L, Krust A, Dupont S, Chambon P, Bayard F, Arnal JF. Estradiol accelerates reendothelialization in mouse carotid artery through a-, but not b-, estrogen receptor. Circulation 2001; 103 : 423–8. [Google Scholar]
- Spyridopoulos I, Sullivan AB, Kearney M, Isner JM, Losordo DW. Estrogen-receptor-mediated inhibition of human endothelial cell apoptosis. Estradiol as a survival factor. Circulation 1997; 95 : 1505–14. [Google Scholar]
- Gourdy P, Mallat Z, Castano C, et al. The atheroprotective effect of 17 -estradiol is not altered in P-selectin- or ICAM-1-deficient hypercholesterolemic mice. Atherosclerosis 2003; 166 : 41–8. [Google Scholar]
- Hodgin JB, Krege JH, Reddick RL, Korach KS, Smithies O, Maeda N. Estrogen receptor alpha is a major mediator of 17beta-estradiol’s atheroprotective effects on lesion size in Apoe−/− mice. J Clin Invest 2001; 107 : 333–40. [Google Scholar]
- Hodgin J, Maeda N. Estrogen and mouse models of atherosclerosis. Endocrinology 2002; 143 : 4495–501. [Google Scholar]
- Dansky H, Charlton S, McGee Harper M, Smith J. T and B lymphocytes play a minor role in atherosclerotic plaque formation in the apolipoprotein E-deficient mouse. Proc Natl Acad Sci USA 1997; 94 : 4642–6. [Google Scholar]
- Elhage R, Clamens S, Reardon-Alulis C, et al. Loss of the atheroprotective effect of estradiol in immunodeficient mice. Endocrinology 2000; 141 : 462–4. [Google Scholar]
- Chiba H, Chambon P, Metzger D. F9 embryonal carcinoma cells engineered for tamoxifen-dependent Cre-mediated site-directed mutagenesis and doxycycline-inducible gene expression. Exp Cell Res 2000; 260 : 334–9. [Google Scholar]
- Elhage R, Maret A, Pieraggi MT, Thiers JC, Arnal JF, Bayard F. Differential effects of interleukin-1 receptor antagonist and tumor necrosis factor binding protein on fatty-streak formation in apolipoprotein E-deficient mice. Circulation 1998; 97 : 242–4. [Google Scholar]
- Mallat Z, Besnard S, Duriez M, et al. Protective role of interleukin-10 in atherosclerosis. Circ Res 1999; 85 : E17–24. [Google Scholar]
- Elhage R, Clamens S, Besnard S, et al. Involvement of interleukin-6 in atherosclerosis but not in the prevention of fatty streak formation by 17beta-estradiol in apolipoprotein E-deficient mice. Atherosclerosis 2001; 156 : 315–20. [Google Scholar]
- Maret A, Coudert JD, Garidou L, et al. Estradiol enhances primary antigen-specific CD4 T cell responses and Th1 development in vivo. Essential role of estrogen receptor alpha expression in hematopoietic cells. Eur J Immunol 2003; 33 : 512–21. [Google Scholar]
- Libby P, Schoenbeck U, Mach F, Selwyn AP, Ganz P. Current concepts in cardiovascular pathology : the role of LDL cholesterol in plaque rupture and stabilization. Am J Med 1998; 104 : S14–8. [Google Scholar]
Liste des figures
|
Figure 1. Les différentes étapes de la constitution de la strie lipidique et de la plaque d’athérosclérose. La première étape est la pénétration de lipoprotéines athérogènes, en particulier des lipoprotéines de faible densité (LDL) à travers la monocouche de cellules endothéliales. L’oxydation des LDL dans l’espace sous-endothélial représente probablement une modification nécessaire aux étapes ultérieures, car les LDL oxydées induisent à leur tour une activation de l’endothélium, en particulier l’expression de molécules d’adhérence pour les leucocytes. Ces molécules d’adhérence endothéliales sont nécessaires pour ralentir les monocytes circulants, les arrêter et permettre leur migration dans l’intima. Les monocytes sont alors activés en macrophages (Ma) ce qui contribue probablement à accroître l’oxydation des LDL (flèches pointillées). Les LDL ainsi davantage oxydées (oxLDL) peuvent être reconnues par des récepteurs éboueurs ( ) (scavenger) présents sur ces macrophages. En fonction de nombreux facteurs systémiques comme les facteurs de risques classiques (hypertension artérielle, hypercholestérolémie, tabagisme, diabète) et de facteurs locaux (degré de dysfonctionnement endothélial, chimiokines, cytokines produites par les différents acteurs cellulaires [endothélium, monocytes-macrophages, cellules musculaires lisses mais aussi lymphocytes]), les macrophages nettoient l’intima et préviennent ainsi l’accumulation de LDL oxydées, ou s’accumulent et deviennent progressivement spumeux (Mas), constituant progressivement la strie lipidique. L’athérome est ainsi à la fois une pathologie métabolique et inflammatoire. L’extension de la strie lipidique tend à être limitée par une réaction cicatricielle des cellules musculaires lisses (CML) de la média qui migrent dans l’intima et sécrètent du collagène (Co). Le niveau d’inflammation de la strie lipidique et la solidité de la chape fibro-musculaire conditionnent la stabilité de la plaque d’athérosclérose, dont la rupture expose des matériaux thrombogènes à l’origine de la formation d’un thrombus, qui menace le territoire artériel en aval en cas d’occlusion de la lumière vasculaire. |
| Dans le texte | |
|
Figure 2. Constructions utilisées dans deux lignées transgéniques pour invalider le gène du récepteur des œstrogènes (ERa). A. Le gène ERa (ERa-WT) comporte 8 exons. Deux codons initiateurs ATG1 (dans l’exon 1) et ATG2 (dans l’exon 2) peuvent être utilisés, à l’origine d’isoformes de longueurs différentes. La partie codante du gène est précédée par une région 5’ non traduite (5’UTR). La première construction d’inactivation de ERa (ERa-Néo KO [10]) a consisté à insérer une cassette néomycine dans le premier exon du gène ERa. La deuxième construction d’inactivation de ERa (ERa-D2 KO [11]) a consisté à exciser le deuxième exon du gène ERa codant pour le domaine de liaison du récepteur à l’ADN. B. Le récepteur des œstrogènes a, ERa (66) comporte 6 domaines (de A à F), dont des domaines de liaison à l’ADN, à l’hormone œstrogène et des fonctions transactivatrices AF1 et AF2. La construction de la lignée ERa-D2 KO ne permet l’expression d’aucune protéine fonctionnelle en l’absence de domaine de liaison à l’ADN. En revanche, nous avons mis en évidence, chez les souris ERa-Néo KO, à la faveur de mécanismes d’épissage alternatif, l’existence de deux formes tronquées de récepteurs : une isoforme de 46 kDa (46) et une molécule chimère de 55 kDa (55) contenant 7 acides aminés de la cassette néomycine. Ces deux récepteurs sont certes dépourvus de la fonction transactivatrice AF1, mais conservent la fonction transactivatrice AF2. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.