")
")
| Issue |
Med Sci (Paris)
Volume 18, Number 6-7, Juin–Juillet 2002
|
|
|---|---|---|
| Page(s) | 697 - 708 | |
| Section | M/S Revues : Articles de Synthèse | |
| DOI | https://doi.org/10.1051/medsci/20021867697 | |
| Published online | 15 juin 2002 | |
Le diagnostic : de la neuropathologie à l’imagerie cérébrale
Diagnosis: from neuropathology to neuroimagery
1
Inserm U.350, Institut Curie, Centre universitaire, Laboratoire 112, 91405 Orsay Cedex, France
2
Service de Neuroradiologie Fischgold, Bâtiment Babinski, Hôpital de la Salpêtrière, 47, boulevard de l’Hôpital, 75651 Paris Cedex 13, France
3
Laboratoire R. Escourolle, Hôpital de la Salpêtrière, 47, boulevard de l’hôpital, 75651 Paris Cedex 13, France
Résumé
Les seuls critères qui permettent, encore aujourd’hui, de porter avec certitude le diagnostic de maladie d’Alzheimer reposent sur l’observation microscopique d’une pathologie neurofibrillaire et de dépôts de peptide Aβ dans le cerveau. Ces lésions sont associées à d’autres altérations telles que des pertes neuronales et synaptiques ou une activation microgliale. Les dépôts de peptide Aβ ont une topographie diffuse. La pathologie neurofibrillaire est répartie de façon « hiérarchique » : sa progression dans les différentes aires corticales suit un ordre chronologique qui permet de déterminer le stade de la maladie. Cette progression peut prendre plusieurs décennies. Détecter précocement les signes de la maladie par neuro-imagerie permettrait de prévoir cette évolution et de tenter de la retarder ou de l’arrêter. Chez l’homme, les études cherchant à visualiser directement les lésions de la maladie par neuro-imagerie étant encore infructueuses, ce sont les signes secondaires qui sont utilisés pour suggérer un diagnostic. L’atrophie temporale médiane, qui peut être détectée par imagerie par résonance magnétique nucléaire, est très évocatrice surtout si elle évolue rapidement lors d’examens répétés. D’autres signes mis en évidence par la résonance magnétique nucléaire (modifications de la diffusion de l’eau, altérations spectroscopiques, réduction de la perfusion cérébrale), la tomographie par émission de positons (TEP) ou la tomographie par émission de photon unique (TEMP) (modification de la perfusion cérébrale et du métabolisme cérébral du glucose, niveau d’activité de certains neurotransmetteurs) sont actuellement en cours d’investigation et pourraient, dans le futur, apporter des informations utiles pour le clinicien dans sa pratique journalière.
Abstract
At the present time, the definite diagnosis of Alzheimer’s disease relies on the microscopic observation of neuro-fibrillary pathology and Aβ peptide deposition in the brain. These lesions are associated with other alterations such as neuronal and synaptic loss or microglial activation. The Aβ deposits are diffusely distributed in the cerebral cortex; the neurofibrillary changes have a hierarchical distribution. The progression of the neurofibrillary pathology in the cortical areas follows a stereo-typed sequence that may help to grade the severity of the disease. Progression may take decades. Being able to detect Alzheimer’s disease related alterations by neuroimagery in the early stage of the disease would be useful to predict its progression and delay its occurrence. In humans, attempts to directly visualize the lesions by neuroimaging techniques are still unsuccessful. Secondary signs have to be used to suggest the diagnosis. Atrophy of the medial temporal lobe and its rapid progression over serially acquired magnetic resonance imaging scans are considered presently the most reliable signs of the disease. Other signs shown by nuclear magnetic resonance (water diffusion alterations, spectroscopic changes, cerebral perfusion reduction), by positron emission tomography (PET) or single photon emission computed tomography (SPECT) (changes in cerebral perfusion and glucose metabolism, or in neurotransmitters activity levels) are currently being evaluated. They might be useful to the clinician in the future.
© 2002 médecine/sciences - Inserm / SRMS
Pourra-t-on, un jour, se passer du microscope pour porter le diagnostic de maladie d’Alzheimer ? En plus des données cliniques, ce diagnostic repose, aujourd’hui encore, surla présence dans le cortex cérébral de marqueurs neuropathologiques dont la taille est schématiquement d’une vingtaine de micromètres (µm). Ces lésions sont le plus souvent observées sur des échantillons post-mortem. Les systèmes optiques doivent les agrandir au minimum d’un facteur 50 pour que leur image à l’œil mesure environ 1 mm, c’est-à-dire soit visible. Une lésion doit recouvrir 2 à 4 pixels pour qu’elle puisse être détectée dans une image numérique ; pour une lésion de 20 µm, chaque pixel doit donc correspondre à 20/4 = 5 µm. Cette résolution de 5 µm, requise pour mettre en évidence les lésions de la maladie d’Alzheimer n’a pas encore été atteinte avec les méthodes non invasives utilisées chez l’homme. L’imagerie d’aujourd’hui utilise des signes secondaires pour suggérer un diagnostic. Des examens complémentaires tels que l’examen du liquide céphalorachidien, l’électroencéphalogramme ou le scanner (dont l’utilisation, récemment remise en question, ne sera pas le sujet de cet article) contribuent aussi au diagnostic, en pratique surtout par l’exclusion d’autres causes de démence.
Lésions microscopiques de la maladie d’Alzheimer
Les lésions microscopiques de la maladie d’Alzheimer peuvent être classées en trois catégories : la pathologie neurofibrillaire, les dépôts de peptide amyloïde Aβ, et les pertes (neuronales ou synaptiques) [1]. La plaque sénile est une lésion composite, comprenant à la fois un dépôt (le cœur) et des lésions neurofibrillaires (la couronne). Les lésions microscopiques sont présentes en grande quantité dans l’isocortex cérébral et l’hippocampe des patients. Elles restent longtemps asymptomatiques, probablement pendant plus d’une décennie, et sont souvent associées à des modifications macroscopiques telles qu’une atrophie cérébrale.
La pathologie neurofibrillaire
Au cours de la maladie d’Alzheimer, des fibrilles anormales, visibles dans le corps cellulaire du neurone, constituent la « dégénérescence neurofibrillaire » (DNF). Des prolongements anormalement dilatés sont aussi constatés dans les plaques séniles dont ils forment la « couronne » . L’espace qui sépare les corps cellulaires des neurones - le neuropile - est traversé de prolongements anormalement courts : les « fibres tortueuses » (neuropil threads), qui sont, pour la plupart, des dendrites [2]. L’observation en microscopie électronique des DNF, de la couronne des plaques séniles et des fibres tortueuses révèleque toutes ces altérations ont le même aspect ultrastructural : chacune est en effet constituée par une paire de filaments disposés en hélice (PHF pour paired helical filaments) ou, comme il a été montré plus récemment, par un ruban torsadé [3]. Les anticorps dirigés contre les protéines tau marquent spécifiquement la pathologie neurofibrillaire [4], dont elles semblent constituer l’élément principal. Des précipités analogues aux PHF peuvent être obtenus in vitro à partir de solutions de protéines tau purifiées en présence de polyanions comme l’héparane sulfate. Enfin, la migration électrophorétique des protéines tau provenant de préparations de DNF isolées est différente de celle obtenue à partir de prélèvements cérébraux normaux [5].
Les dépôts de peptide amyloïde
Le centre de la plaque sénile comporte un dépôt d’environ 10 à 20 µm de diamètre (Figure 1). Celui-ci a d’abord été pris pour de la nécrose - justifiant le terme de nécrose miliaire qui lui a été appliqué autrefois. La nature « amyloïde » du dépôt a été affirmée par la coloration au rouge Congo - qui se fixe sélectivement sur les substances amyloïdes et induit une biréfringence verte en lumière polarisée - et par l’examen en microscopie électronique qui révèle sa nature fibrillaire. Des dépôts amyloïdes se produisent dans la paroi des artères et des veines, méningées ou parenchymateuses, ainsi que des capillaires. La prévalence de cette angiopathie amyloïde au cours de la maladie d’Alzheimer est diversement appréciée : elle est en tout cas fréquente, sinon constante [6]. Les substances amyloïdes ont en commun leur structure chimique, en feuillets β plissés, qui les rend insolubles. Elles sont constituées du peptide amyloïde Aβ, de 40 à 42 acides aminés. Les anticorps dirigés contre ce peptide marquent le centre des plaques séniles et les dépôts vasculaires de l’angiopathie amyloïde. Ils mettent aussi en évidence une lésion jusqu’alors invisible ou sous-estimée : les dépôts diffus de peptide Aβ, beaucoup plus volumineux que les plaques séniles, plus irréguliers et moins bien limités [7]. Ces dépôts diffus sont dépourvus de la couronne de prolongements nerveux dégénérés propres aux plaques séniles. L’utilisation d’anticorps spécifiques de l’extrémité C-terminale du peptide Aβ a permis de montrer que le peptide de 42 acides aminés est localisé préférentiellement dans les dépôts diffus et celui de 40 acides aminés sur les dépôts focaux, en particulier le cœur des plaques séniles [8].
 |
Figure 1. Aspect des lésions observées dans la maladie d’Alzheimer à l’examen microscopique. Toutes les photographies ont été prises avec un objectif x100 ; le trait de l’image A représente 20 µm. A. Plaque sénile ; immunohistochimie du peptide Aβ. Un dépôt dense de peptide Aβ (grande flèche) occupe le centre de la plaque sénile. Elle est entourée d’une couronne claire (correspondant aux prolongements nerveux de l’image C), puis d’un halo de dépôt diffus (petites flèches). Un noyau de macrophage est marqué par une tête de flèche. B. Plaque sénile colorée par le rouge Congo, colorant qui se fixe sélectivement sur les substances amyloïdes. La partie amyloïde de la plaque sénile (correspondant au centre dense de la photo A) est indiquée par une flèche. La tête de flèche marque le noyau d’un macrophage. C. Plaque sénile ; immunohistochimie de la protéine tau. Le centre de la plaque (grande flèche) est à peine visible. Il est entouré de prolongements nerveux dégénérés, marqués par l’anticorps anti-tau (petites flèches). Le noyau de deux macrophages est indiqué par des têtes de flèches. D. Dégénérescence neurofibrillaire ; immunohistochimie de la protéine tau. Deux neurones sont visibles (marqués a et b). Le noyau est indiqué par une grosse tête de flèche, le nucléole par une petite tête de flèche. La dégénérescence neurofibrillaire est constituée par des fibrilles de protéine tau dans le corps cellulaire du neurone a (flèches). |
La plaque sénile : interface entre la pathologie neurofibrillaire et les dépôts
La plaque sénile comprend un dépôt focal de peptide Aβ, situé en son cœur, et une couronne de prolongements chargés en protéine tau. Le terme de « plaque neuritique » est parfois utilisé pour insister sur la présence de ces prolongements et est opposé au terme de « plaque primitive », qui désigne les lésions uniquement faites de dépôt de peptide Aβ. La nature des prolongements de la couronne a été discutée. Les observations en microscopie électronique ont révélé l’origine principalement axonale de ces prolongements et une faible participation dendritique [9]. Certains prolongements dystrophiques observés aux stades précoces de la plaque sénile ne comportent pas d’épitope de la protéine tau, mais sont marqués par des anticorps dirigés contre l’APP (le précurseur du peptide amyloïde) et contre l’ubiquitine [10].
Des cellules microgliales activées, des composants précoces de la cascade du complément, l’α-antichymotrypsine, différents types d’interleukine ont été identifiés au sein de la plaque sénile et témoignent d’un processus inflammatoire persistant [11]. La microglie activée pourrait jouer un rôle dans la résorption de la substance amyloïde ou, selon une autre hypothèse, dans sa production. Enfin, plusieurs composants de la matrice extracellulaire, tels que ICAM-1 (intercellular adhesion molecule), la thrombospondine, les protéoglycanes à héparane sulfate, s’accumulent dans la plaque sénile.
Les pertes neuronale et synaptique
La perte neuronale
La perte neuronale, naguère considérée comme un des signes principaux de la maladie, est en réalité difficile à quantifier. Elle est liée à la présence de DNF [12] : elle est donc tardive dans les régions où la DNF n’apparaît qu’aux derniers stades de la maladie. Elle est précoce au contraire dans les régions les plus touchées par la DNF comme le cortex entorhinal [13]. Après la mort du neurone, la DNF subsiste - comme un « fantôme » (selon l’expression anglaise ghost tangle). De nombreux « fantômes » sont observés dans l’hippocampe ou l’aire entorhinale. La somme des fantômes et des neurones survivants est proche du nombre total de neurones observés chez les sujets normaux [14].
Le rôle de l’apoptose dans la perte neuronale a été proposé mais est encore discuté. S’il a été montré que la fragmentation de l’ADN dans les neurones était fréquente au cours de la maladie d’Alzheimer, il est difficile d’envisager que tous les neurones comportant de l’ADN fragmenté soient destinés à mourir en quelques heures ou quelques jours : la maladie devrait alors avoir une évolution aiguë (et non chronique). De plus, la migration en échelle de l’ADN nucléaire, une anomalie typique de l’apoptose, n’a pu être mise en évidence, pas plus que d’autres stigmates morphologiques caractéristiques. Cependant, l’activation de la caspase 3 est observée dans quelques neurones témoignant d’une apoptose qui ne concerne cependant qu’un petit nombre de cellules, plus compatible avec une évolution chronique [15]. Selon une autre hypothèse, les phénomènes apoptotiques pourraient être limités à la synapse.
(→) m/s 1993, n°6-7, p. 802 et 1993, n°8-9, p. 1000
La perte synaptique
Les premiers résultats obtenus par dénombrement des synapses en microscopie électronique ont mis en évidence une diminution du nombre des synapses mais une augmentation de leur surface d’apposition [16]. L’analyse de protéines synaptiques a montré que les composants membranaires (synaptotagmine, SNAP-25 et syntaxine 1/HPC-1) sont peu diminués tandis que les composants vésiculaires (synaptobrévine et synaptophysine) le sont beaucoup plus (→) [17]. Il existe une baisse marquée et précoce de la synaptophysine dont la corrélation avec la détérioration intellectuelle est discutée : elle est jugée excellente par certains [18], qui en font le meilleur corrélat de la démence ou, au contraire, inférieure à celle qui est observée pour les DNF par d’autres [19].
Les corrélations clinico-pathologiques
L’analyse prospective de groupes de sujets âgés a permis d’établir des corrélations entre la densité des lésions neurologiques et la détérioration intellectuelle [19]. Les corrélations sont bonnes avec la pathologie neurofibrillaire qu’il s’agisse de la densité des DNF ou des plaques neuritiques (c’est-à-dire comportant une couronne de prolongements chargés de PHF). Les dépôts diffus de peptide Aβ à eux seuls ne provoquent pas de détérioration intellectuelle ou seulement des troubles minimes [20]. La signification de ce syndrome clinico-pathologique caractérisé par d’abondants dépôts diffus sans altération intellectuelle, est encore débattue : il pourrait s’agir d’un vieillissement anormal ou d’un stade précoce d’une maladie d’Alzheimer débutante [21].
La progression des lésions : description et mécanisme
Diffusion des dépôts de peptide Aβ
La topographie des dépôts de peptide Aβ et celle de la pathologie neurofibrillaire sont différentes. Les dépôts amyloïdes intéressent, dès les stades précoces de la maladie, la plupart des aires du cortex cérébral, y compris celles qui sont dépourvues de pathologie neurofibrillaire. Dans des cas de trisomie 21 où le décès est survenu à un âge précoce, des dépôts de peptide Aβ ont été observés, avant toute pathologie neurofibrillaire, un argument de poids en faveur de l’hypothèse de la « cascade amyloïde » selon laquelle l’accumulation de peptide Aβ provoquerait la dégénérescence neurofibrillaire et la perte neuronale [22]. Cette hypothèse ne tient pas compte, cependant, de l’observation selon laquelle la pathologie fibrillaire précède, dans la population générale, les dépôts de peptide Aβ [23].
Hiérarchie lésionnelle dans la pathologie neurofibrillaire
L’étude de la pathologie neurofibrillaire de patients décédés à différents stades de la maladie et de sujets âgés considérés comme intellectuellement normaux a permis de définir un ordre dans lequel apparaissent les lésions (Figure 2). Chez les sujets âgés normaux, on observe fréquemment des DNF dans l’aire entorhinale, une région située entre l’isocortex et l’hippocampe, et sur laquelle convergent les voies issues des aires associatives multimodales. Dans certains cas de troubles mnésiques isolés, des DNF abondantes sont trouvées dans l’hippocampe, et sont toujours associées à celles du cortex entorhinal. Enfin, chez les patients déments, les lésions neurofibrillaires sont observées dans l’isocortex, et sont alors présentes aussi dans l’hippocampe et le cortex entorhinal. Les régions corticales touchées le sont donc dans un ordre stéréotypé [24, 25] (cortex entorhinal, hippocampe, isocortex d’abord, puis les aires associatives et, tardivement, les aires primaires), correspondant à une « hiérarchie » lésionnelle qui témoigne d’une progression temporelle de la pathologie neurofibrillaire. Dans les aires entorhinale et hippocampique, les lésions neurofibrillaires initiales sont associées ensuite aux dépôts de peptide Aβ et, plus tardivement, à la perte neuronale, source d’atrophie. Dans les aires isocorticales, ce sont d’abord les dépôts diffus qui apparaissent, suivis des altérations neurofibrillaires et de la perte neuronale. Dans tous les cas, l’atrophie détectée par les méthodes d’imagerie in vivo est donc un phénomène tardif.
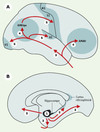 |
Figure 2. Représentation schématique de la progression de la pathologie neurofibrillaire dans le cortex cérébral. A. Vue latérale. B. vue médiale. La première aire corticale à être affectée par les dégénérescences neurofibrillaires est le cortex entorhinal (marqué 1). A l’étape suivante, les lésions sont observées dans l’hippocampe (flèche 2) et le cortex rétrosplénial. La pathologie neurofibrillaire intéresse ensuite la partie antérieure du lobe temporal (flèche 3). Les aires associatives uni- puis multimodales frontale (AMAfr) et temporo-pariéto-occipitales (AMAtpo) sont concernées à l’étape suivante (flèches marquées 4). Les aires primaires (motrice = M1, somesthésique = S1, auditive = A1, visuelle = V1) sont atteintes en dernier lieu (flèches 5). (D’après [24, 25, 59]). |
Imagerie des lésions microscopiques de la maladie d’Alzheimer
Expérimentalement, chez une souris transgénique APP développant des dépôts amyloïdes, il est possible de visualiser les plaques in vivo grâce à une technique de microscopie multiphoton qui « regarde » le cerveau à travers une fenêtre faite en rabotant le crâne jusqu’à ce qu’il soit transparent. Les plaques sont marquées par un fluorophore (thioflavine S) injecté dans le cerveau. Le marqueur se fixe sur les dépôts amyloïdes et est révélé par une excitation par de multiples photons à faible énergie, qui n’altèrent donc pas les neurones. Cette méthode, dont la résolution spatiale est de l’ordre de 1 µm, permet de révéler des plaques qui se trouvent jusqu’à 150 µm sous la surface du cortex [26]. Elle permet de suivre longitudinalement leur évolution mais reste trop invasive pour être applicable à l’homme. La détection des lésions microscopiques de la maladie d’Alzheimer, in vivo, par des méthodes non invasives, constituerait un progrès décisif : le diagnostic « certain » pourrait être fait du vivant du malade et il deviendrait possible de suivre l’évolution de la pathologie. Plusieurs études in vitro, en microscopie-IRM (imagerie par résonance magnétique nucléaire), ont cherché à évaluer les mécanismes de contraste associés aux plaques séniles, mais les différents protocoles utilisés (contrastes T1, T2 ou en diffusion) n’ont pas réussi à les visualiser [27]. Des études de faisabilité ont suggéré que les plaques séniles, contenant du fer (paramagnétique), doivent induire un effet de sensibilité sur des images RMN pondérées en T2* sur lesquelles elles devraient apparaître en faible intensité [28]. De telles images ont, de fait, été récemment interprétées comme celles de grosses plaques séniles (plus de 100 µm de diamètre) [27]. Ce résultat n’a pas été confirmé et il semblerait, qu’au mieux, seule une proportion infime des plaques séniles (les plus grosses) puisse être directement détectée en RMN [28] (Figure 3). D’autres stratégies sont actuellement en cours d’investigation qui permettraient de visualiser directement les plaques séniles en IRM [29], tomographie par émission de positons (TEP) ou tomographie par émission de photon unique (TEMP) [30]. Elles reposent sur le développement de marqueurs qui, après injection par voie intraveineuse, passent la barrière hémato-encéphalique puis se fixent sur les plaques séniles avant d’être détectés par ces techniques de neuro-imagerie.
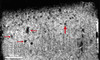 |
Figure 3. Image obtenue par l’analyse en microscopie-IRM d’échantillon de tissus d’un patient atteint de maladie d’Alzheimer. Cette image, pondérée en T2*, a une résolution isotrope de 23,4 µm par voxel. Les structures punctiformes hypo-intenses (flèche rouge épaisse) ont été interprétées comme des plaques séniles [27]. Une étude plus récente suggère qu’il s’agirait de sections de vaisseaux, tout comme les structures plus allongées (flèches rouges fines) [28]. |
Altérations macroscopiques associées à la maladie d’Alzheimer : l’atrophie cérébrale
L’atrophie cérébrale se produisant lors de la maladie d’Alzheimer induit une dilatation du système ventriculaire, un agrandissement des espaces périvasculaires et un élargissement des sillons corticaux. Elle peut être appréciée par la mesure du poids ou du volume du cerveau à l’examen post-mortem ou par celle du volume cérébral par les techniques d’imagerie cérébrale. Ces mesures entraînent cependant des difficultés car elles sont modifiées en particulier par l’état d’hydratation cérébrale (notamment pour les mesures post-mortem). Les mesures directes, effectuées en postmortem, sont fortement corrélées à celles qui ont été faites sur les images RMN, également post-mortem. Nous ne connaissons pas d’étude qui ait comparé systématiquement les images obtenues in vivo et les constatations post-mortem.
Poids ou volume du cerveau
Le poids du cerveau diminue au cours de la maladie d’Alzheimer (200 à 300 g, soit 15 à 25 % selon la plupart des auteurs), surtout si elle est de début précoce, la perte étant en moyenne inférieure à 10 % lorsque le début est tardif. Le poids du cerveau diminue aussi avec l’âge mais de façon beaucoup moins marquée : constant jusqu’à l’âge de 50-55 ans, il perd environ 2 % de sa valeur tous les 10 ans [31]. À l’échelon individuel, le chevauchement des valeurs normales et pathologiques est tel qu’on ne peut en tirer d’informations diagnostiques sinon dans les cas d’atrophie importante (celle-ci est alors assez spécifique de la maladie d’Alzheimer). Enfin, la perte de poids affecte différemment les lobes cérébraux au cours de la maladie : 41 %, 30 % et 14 % (non significatif) respectivement pour les lobes temporal, pariétal et frontal [32].
Atrophie corticale et dilatation ventriculaire
L’atrophie corticale se produisant lors de la maladie d’Alzheimer est principalement liée à la réduction de la longueur du ruban cortical, celle-ci étant elle-même la conséquence de la perte de neurones et de fibres disposés en colonnes perpendiculaires à la surface corticale. La réduction de la longueur corticale est corrélée aux scores cognitifs [33]. Les mesures de l’atrophie corticale globale ou d’élargissement ventriculaire, évaluées in vivo par scanner ou IRM, ont une sensibilité diagnostique faible (46 ± 20 %) mais une bonne spécificité (90 ± 7 %) en raison du large chevauchement avec les altérations liées au vieillissement normal. Ainsi, un volume cérébral normal pour l’âge du sujet est considéré comme compatible avec le diagnostic de maladie d’Alzheimer [34].
L’atrophie corticale n’est pas homogène, elle prédomine sur les aires parasagittales et le lobe temporal qui est la seule région atrophiée lorsque la maladie d’Alzheimer débute tardivement (après 80 ans) [35]. L’atrophie temporale interne, en particulier hippocampique et parahip-pocampique, peut être détectée par les méthodes modernes de neuro-imagerie (Figures 4 et 5). L’atrophie hippocampique est de 30 à 50 % dans les formes modérées de la maladie, 20 à 30 % dans les formes légères et environ 10 % dans les formes débutantes. La mise en évidence d’une atrophie temporo-hipoccampique permet donc non seulement de diagnostiquer la maladie avec une sensibilité de 80 à 100 % et une spécificité de 80 à 95 % selon les études [36], mais aussi de prévoir l’apparition d’une démence chez des sujets âgés qui présentent des troubles cognitifs légers [37]. Elle est statistiquement liée aux performances cognitives dans les cas de démence d’Alzheimer avérée [38]. Inversement, l’absence d’atrophie de la partie interne du lobe temporal est un argument pour éliminer le diagnostic [39]. L’atrophie hippocampique peut être observée dans d’autres affections, souvent associées à la maladie d’Alzheimer, comme la maladie de Parkinson, surtout si elle s’accompagne de démence, et la démence à corps de Lewy [39]. Elle peut aussi être présente dans les démences vasculaires, lorsqu’il existe, par exemple, une ischémie du territoire de l’artère cérébrale postérieure ou dans des cas de sclérose hippocampique. Étant donné que, lors de la maladie d’Alzheimer, les DNF entorhinales précèdent celles de l’hippocampe [25], des études en IRM ont cherché à déterminer si la mesure d’une atrophie entorhinale permettait un diagnostic plus précoce de la maladie et ont conclu par la négative. Enfin, l’atrophie de l’amygdale, qui pourrait précéder celle de l’hippocampe, a aussi été proposée comme un marqueur précoce [40].
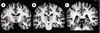 |
Figure 4. Anatomie hippocampique par IRM. Coupes coronales obliques perpendiculaires au grand axe des formations hippocampiques passant par l’uncus (A), le corps (B) et la queue (C) de l’hippocampe. Les formations hippocampiques sont indiquées par les flèches rouges, l’amygdale par la flèche jaune. |
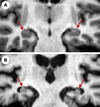 |
Figure 5. Atrophie hippocampique dans la maladie d’Alzheimer. Coupe coronale pondérée en T1 passant par le corps des formations hippocampiques (flèches) d’un sujet témoin ( A ) et d’un patient atteint de maladie d’Alzheimer ( B ). Il existe une atrophie importante des formations hippocampiques du patient atteint de maladie d’Alzheimer. |
Progression de l’atrophie cérébrale
La rapidité de progression distingue clairement l’atrophie corticale de la maladie d’Alzheimer de celle du vieillissement « normal », d’où l’intérêt des méthodes de neuroimagerie non invasives qui permettent un suivi longitudinal des sujets. Une telle étude, réalisée au cours d’une période de 5 à 8 ans chez des personnes ayant un risque génétique de développer une maladie d’Alzheimer, mais qui ne présentaient pas de symptôme en début d’étude, a révélé que la progression rapide de l’atrophie permet de distinguer celles qui développent la maladie (diminution de volume du cerveau de 1,5 % par an) de celles qui ne la développent pas (diminution de volume de 0,1 %) [41]. La vitesse d’évolution de l’atrophie est, de plus, corrélée à la progression des déficits neuropsychologiques. L’augmentation annuelle du volume des ventricules latéraux permet aussi de détecter la maladie d’Alzheimer avec une sensibilité et une spécificité proche de 100 %. L’aggravation de l’atrophie en 1 ou 2 ans a donc été retenue comme un signe de suspicion de la maladie [34]. Les difficultés techniques rencontrées dans la comparaison des images d’un même cerveau à plusieurs années d’intervalle sont en cours de résolution grâce à des procédés de déformation et de recalage d’images qui permettent, grâce à divers algorithmes mathématiques, de superposer des images réalisées à des moments différents même si la position du patient, et donc celle des images, varient légèrement [41]. Ces procédés devraient faciliter le suivi des patients en imagerie.
Altérations de la substance blanche
Les neurones corticaux, dont les corps cellulaires et les dendrites sont localisés dans la substance grise, peuvent se projeter à distance (par exemple dans une autre région corticale) en traversant la substance blanche. La perte de ces axones, à la suite de la mort du neurone, provoque donc une diminution du volume de la substance blanche. La constance du rapport substance blanche/substance grise au cours de la maladie d’Alzheimer suggère que la perte du corps cellulaire et des dendrites du neurone provoque une perte de volume grossièrement égale à celle causée par la disparition de son axone. Les fibres du corps calleux relient des régions corticales homologues : les lobes frontaux pour sa partie rostrale, les lobes temporaux pour sa partie médiane, et les lobes pariétaux, entre autres, pour sa partie caudale (splenium). L’atrophie des différentes parties du corps calleux a pu être corrélée, en IRM, avec la diminution de volume des lobes correspondants. Comme les régions atrophiées du cerveau varient selon le type de démence, la localisation des zones du corps calleux atrophiées pourrait aider au diagnostic différentiel de diverses démences (maladie d’Alzheimer, paralysie supra-nucléaire progressive, démence fronto-temporale) [42].
Les altérations de la substance blanche peuvent aussi être étudiées grâce à des protocoles d’IRM de diffusion qui évaluent le mouvement aléatoire des molécules d’eau dans un tissu. On peut en effet déterminer un coefficient de diffusion apparent de l’eau (ADC pour apparent diffusion coefficient) selon une direction de l’espace déterminée par l’orientation d’un gradient de champ magnétique dit « de diffusion » inséré dans la séquence d’imagerie. En combinant plusieurs mesures d’ADC selon des orientations différentes, on peut explorer les mouvements de diffusion des molécules d’eau dans les trois dimensions de l’espace et déterminer s’ils sont isotropes (dans toutes les directions) ou anisotropes (avec une orientation préférentielle). L’anisotropie de la diffusion de l’eau est présente dans les tissus possédant une microstructure orientée, en particulier la substance blanche. Elle est moins marquée chez les patients atteints de maladie d’Alzheimer, une diminution qui reflèterait des modifications microstructurales (perte de l’orientation des fibres) associées à une diminution, consécutive à l’atrophie corticale, des connexions entre les aires [43]. Des différences ont aussi été trouvées dans l’hippocampe entre des sujets sains et des sujets présentant des altérations cognitives légères [44]. D’autres études sont cependant nécessaires pour préciser l’intérêt de l’IRM de diffusion dans le diagnostic précoce de la maladie d’Alzheimer.
Spectroscopie RMN
La spectroscopie par RMN (SRM) du proton (1H) permet la mesure non invasive d’un certain nombre de métabolites cérébraux. Il a ainsi été montré qu’au cours de la maladie d’Alzheimer, la concentration en N-acétyl-aspartate (NAA) diminue tandis que celle du myo-inositol augmente dans les régions occipitale, temporale, pariétale et frontale [45]. Dans certaines études, ces modifications ont été corrélées avec l’état cognitif des patients et l’atrophie cérébrale. L’origine de ces altérations est encore discutée : la diminution de la concentration en N-acétyl-aspartate refléterait une diminution de la viabilité cellulaire et le myo-inositol pourrait être un marqueur glial plutôt d’origine membranaire. La sensibilité diagnostique de la méthode est élevée, mais sa spécificité est faible [45]. À ce jour, aucune étude prospective n’a, à notre connaissance, été réalisée pour préciser l’intérêt des paramètres spectroscopiques dans l’étude des altérations cognitives légères, et on ne sait pas s’ils aideraient à prévoir l’évolution vers une maladie d’Alzheimer avérée.
D’autres noyaux peuvent être étudiés en spectroscopie RMN. La spectroscopie du phospore (31P) permet d’évaluer des concentrations en phospholipides membranaires, comme les phosphomonoesters et les phosphodiesters, et en éléments phosphatés intervenant dans le métabolisme énergétique comme le phosphate inorganique, la phospho-créatine et les nucléotides tri-phosphates dont l’ATP. Les phosphomonoesters augmentent dans les aires temporo-pariétale et préfrontale des patients atteints de maladie d’Alzheimer débutante et rejoignent la normale dans les cas évolués [46], ce qui suggère une altération précoce du métabolisme membranaire. Les niveaux de phosphocréatine et d’ATP dans les régions préfrontale, frontale et temporo-pariétale sont bas dans la maladie d’Alzheimer débutante et normaux dans les cas évolués [46]. Ces résultats témoignent d’un stress énergétique se produisant lors des phases débutantes de la maladie.
Imagerie fonctionnelle cérébrale
Plusieurs paramètres de l’activité cérébrale, au repos ou lors de la réalisation de tâches cognitives spécifiques, peuvent être mesurés grâce aux techniques d’imagerie fonctionnelle (Tableau I). Le métabolisme cérébral du glucose peut être évalué avec la TEP. La perfusion cérébrale au repos peut être évaluée par TEP (mesure du flux sanguin cérébral), TEMP ou, plus récemment, grâce à des techniques d’IRM fonctionnelle (IRMf)(→). Si perfusion cérébrale et métabolisme sont fortement corrélés, on ne connaît pas bien les mécanismes spécifiques qui les associent. TEP et TEMP évaluent la distribution tissulaire de radio-traceurs injectés ou inhalés, tandis que l’IRMf permet d’obtenir une meilleure résolution spatio-temporelle, ne nécessite pas l’utilisation de radio-traceurs et apporte aussi des informations structurales.
(→) m/s 1999, n°4, p. 447
Techniques d’imagerie cérébrale permettant de réaliser des images fonctionnelles. TEP : tomographie par émission de positons. TEMP : tomographie par émission de photon unique. IRM : imagerie par résonance magnétique.
Au cours du vieillissement normal, l’activité cérébrale au repos est réduite dans le lobe frontal et la substance blanche sous-corticale. Ces modifications sont plus variables dans les aires corticales associatives et le cortex temporal [47]. Au cours de la maladie d’Alzheimer, l’activité cérébrale est particulièrement réduite dans les régions temporale, préfrontale et pariétale et est alors corrélée à l’altération des performances cognitives [48]. L’utilisation des méthodes d’imagerie fonctionnelle pour prédire l’évolution de sujets non déments vers une démence est en cours d’évaluation. Les méthodes mesurant la perfusion cérébrale (par exemple, la TEMP) (→)semblent avoir une faible valeur prédictive de l’évolution de troubles cognitifs mineurs vers la maladie d’Alzheimer et la mesure de la perfusion cérébrale en routine n’est pas très intéressante d’un point de vue diagnostique [49]. Combiner la mesure de la perfusion cérébrale à celle de l’atrophie hippocampique pourrait peut-être augmenter la valeur diagnostique de ces deux méthodes. Les modifications du métabolisme glucidique cérébral au repos semblent avoir une meilleure valeur prédictive de l’évolution du déclin cognitif chez des sujets âgés ayant des troubles de la mémoire légers et permettraient de diagnostiquer la maladie d’Alzheimer de façon plus précoce que l’atrophie cérébrale [50].
(→) m/s 1999, n°4, p. 467
Quel que soit son mode d’évaluation (perfusion cérébrale ou métabolisme cérébral du glucose), l’activation cérébrale est corrélée à l’activité neuronale. Son altération pourrait donc refléter la perte neuronale ou être la conséquence d’autres lésions, par exemple des modifications vasculaires ou l’accumulation de DNF dans les neurones. Les altérations du métabolisme du glucose lorsque le sujet est au repos seraient aussi associées à l’altération du fonctionnement synaptique.
L’activité cérébrale lors de la réalisation de tâches cognitives spécifiques, et en particulier mnésiques, peut aussi être évaluée par imagerie fonctionnelle. Dans ces épreuves, le lobe frontal de sujets âgés normaux semble plus activé que celui des jeunes, ce qui suggère qu’ils utilisent des réseaux neuronaux différents [51]. Certaines études suggèrent que l’activité cérébrale n’est pas modifiée lors de la phase initiale de la maladie d’Alzheimer et que seules les phases tardives de la démence sont associées à un déclin de l’activité cérébrale [52]. D’autres, au contraire, suggèrent qu’il existe une diminution de l’activation hippocampique et parahippocampique chez des patients atteints de maladie d’Alzheimer probable lors de la réalisation d’épreuves mnésiques et une augmentation de l’activité dans les régions corticales qui ne sont pas, ou moins, atteintes par la maladie. Cette diminution d’activité temporale serait retrouvée chez des sujets âgés présentant des troubles de mémoire discrets et isolés. Enfin, chez des sujets normaux, à risque car porteurs de l’allèle ε4 de l’apolipoprotéine E (apoE), des altérations de l’activation cérébrale sont détectables très tôt, avant même que la mémoire ne soit altérée [53].
Déficits neurochimiques et imagerie
L’activité de l’acétylcholine transférase (ChAT) et d’autres marqueurs cholinergiques est abaissée dans le cortex cérébral au cours de la maladie d’Alzheimer. L’importance clinique des altérations cholinergiques a été diversement appréciée. Plusieurs études ont montré une bonne corrélation entre la sévérité de la démence, le déficit en ChAT cortical et les lésions du noyau basal de Meynert où les fibres cholinergiques prennent naissance [54]. Parmi les fonctions cognitives qui reflètent le mieux les lésions du noyau basal de Meynert figure la mémoire. Selon « l’hypothèse cholinergique », la déplétion en acétylcholine est à l’origine de déficits cognitifs (en particulier mnésiques) qu’un traitement adéquat est susceptible d’amender, et améliore en effet. Mais, les lésions sont beaucoup plus diffuses, et les DNF et les pertes neuronales hippocampiques, par exemple, interviennent aussi dans les troubles de la mémoire. Par ailleurs, le déficit neurochimique n’est pas limité à l’acétylcholine. Il touche en fait l’ensemble des grands systèmes de neurotransmission (sérotoninergique, noradrénergique, dopaminergique) dont les corps cellulaires projettent vers le cortex. En outre, dans 80 % des cas de maladie d’Alzheimer et tout particulièrement si elles sont associées à l’allèle ε4 de l’ApoE, il existe un déficit cérébral en somatostatine. Ce déficit pourrait être la conséquence de la présence de DNF dans un ensemble de neurones non-pyramidaux somatostatinergiques des couches II-III et VI du cortex, non colocalisés avec le neuropeptide Y. L’atteinte des autres neuropeptides ou neurotransmetteurs est plus discutée. Si le neuropeptide Y (→), la substance P, la corticolibérine, le glutamate sont légèrement diminués au cours de la maladie d’Alzheimer, la majorité des autres neuropeptides et neurotransmetteurs ne semble pas affectée [55].
Les déficits en neurotransmetteurs, au cours de la maladie d’Alzheimer, sont en outre hétérogènes. Par exemple, les déficits en somatostatine et en acétylcholine ne sont pas corrélés, et il est bien établi que c’est au déficit en acétylcholine que la démence est surtout corrélée [56], même s’il n’est pas détecté chez certains patients. De plus, ces déficits sont, en réalité, variables d’un individu à l’autre. On peut envisager qu’un profil de déficit pourra un jour être établi chez un patient donné grâce aux méthodes récentes de neuro-imagerie. Les techniques de TEP ou de TEMP utilisant des ligands radio-marqués spécifiques de récepteurs de neurotransmetteurs permettent d’évaluer le lieu d’action et l’activité des neurotransmetteurs in vivo. Elles permettent déjà d’évaluer in vivo l’activité cholinergique [57] ou sérotoninergique [58], dont les modifications interindividuelles au cours de la maladie pourraient expliquer certaines des hétérogénéités cliniques, et pourront peutêtre permettre dans le futur d’adapter les traitements substitutifs à chaque patient [57].
(→) m/s 1998, n°8-9, p. 907
Conclusions
La neuropathologie est une étape primordiale dans le diagnostic de certitude de la maladie d’Alzheimer dont les lésions caractéristiques ne sont encore détectables que sur des échantillons biopsiques ou post-mortem. Outre leur rôle diagnostique, les études neuropathologiques permettent d’améliorer la connaissance des altérations cérébrales associées aux lésions de la maladie et de mieux comprendre les cascades de réactions menant aux troubles mnésiques caractéristiques. Actuellement, les techniques de neuro-imagerie ne sont qu’une aide au diagnostic chez des patients qui présentent déjà des symptômes cognitifs, et s’ajoutent aux autres méthodes diagnostiques, dont la neuropsychologie reste le fer de lance. Prédire l’apparition de la démence, chez des patients présentant des troubles cognitifs légers ou chez des sujets à risque d’un point de vue génétique, constitue aujourd’hui le vrai défi de la neuro-imagerie. Le diagnostic précoce permettra, lorsqu’une thérapeutique sera disponible, de traiter les malades avant l’apparition de lésions irréversibles. La recherche de manifestations pré-cliniques nécessite des études lourdes au cours desquelles des sujets, présentant des altérations cognitives mineures, sont suivis longitudinalement pour guetter la modification de paramètres de neuro-imagerie en parallèle avec l’apparition d’une maladie d’Alzheimer. Aucune technique de neuro-imagerie ne permet encore d’accéder à un paramètre ayant un rapport direct, simple, connu, avec les deux lésions caractéristiques de la maladie. La micro-imagerie des plaques séniles a donné des résultats mitigés et n’est pas encore applicable in vivo chez l’homme. Les paramètres étudiés en neuro-imagerie ne sont que le reflet des altérations tardives de la maladie. L’évaluation de l’atrophie cérébrale (temporale interne) et de son évolution, par le scanner et l’IRM, et en utilisant des méthodes précises de recalage, semble la méthode la plus utilisable en clinique. Son rapport intérêt/coût est relativement bon : la simple inspection d’une image scanner ou IRM peut souvent, lorsqu’elle est associée à un examen neuropsychologique classique, apporter suffisamment d’informations au clinicien pour diagnostiquer une maladie d’Alzheimer avec une sensibilité et une spécificité suffisantes. Les méthodes d’imagerie par diffusion et les méthodes spectroscopiques sont en cours d’exploration mais ne sont pas encore reconnues. Des espoirs importants résident dans la possibilité de détecter précocement les altérations de la perfusion cérébrale et du métabolisme cérébral du glucose. Face à l’absence de spécificité suffisante des différents examens de neuro-imagerie, leur utilisation conjointe pourrait aider à affiner la spécificité des méthodes de diagnostic précoce. Cela n’est cependant pas toujours vérifié d’un point de vue clinique et est peu applicable en pratique pour des raisons économiques. Il est possible que la neuro-imagerie, dans un avenir proche, soit utile pour suivre l’évolution des symptômes sous traitement. Les techniques de TEP et TEMP, par exemple, semblent prometteuses pour évaluer, in vivo, des systèmes de neurotransmetteurs spécifiques ce qui pourrait guider, dans le futur, les traitements à visée substitutive.
Références
- Hauw JJ, Duyckaerts C. Alzheimer disease. In : Duckett S, De La Torre JC, eds. Pathology of the aging nervous system, 2nd ed. New York : Oxford University Press, 2001: 207–63. [Google Scholar]
- Duyckaerts C, Kawasaki H, Delaère P, Rainsard C, Hauw JJ. Fiber disorganization in the neocortex of patients with senile dementia of the Alzheimer type. Neuropathol Appl Neurobiol 1989; 15 : 233–47. [Google Scholar]
- Pollanen MS, Markiewicz P, Goh MC. Paired helical filaments are twisted ribbons composed of two parallel and aligned components : image reconstruction and modeling of filament structure using atomic force microscopy. J Neuropathol Exp Neurol 1997; 56 : 79–85. [Google Scholar]
- Brion JP, Passareiro H, Nunez J, Flament-Durand J. Mise en évidence immunologique de la protéine tau au niveau des lésions de dégénérescence neurofibrillaire de la maladie d’Alzheimer. Arch Biol (Brux) 1985; 95 : 229–35. [Google Scholar]
- Delacourte A, Flament S, Dibe EM, et al. Pathological proteins Tau 64 and 69 are specifically expressed in the somatodendritic domain of the degenerating cortical neurons during Alzheimer’s disease. Demonstration with a panel of antibodies against Tau proteins. Acta Neuropathol 1990; 80 : 111–7. [Google Scholar]
- Joachim CL, Duffy LK, Morris JH, Selkoe DJ. Protein chemical and immunocytochemical studies of meningovascular ß-amyloid protein in Alzheimer’s disease and normal aging. Brain Res 1988; 474 : 100–11. [Google Scholar]
- Delaère P, Duyckaerts C, He Y, Piette F, Hauw JJ. Subtypes and differential laminar distributions of ßA4 deposits in Alzheimer’s disease : relationship with the intellectual status of 26 cases. Acta Neuropathol (Berl) 1991; 81 : 328–35. [Google Scholar]
- Barelli H, Lebeau A, Vizzavona J, et al. Characterization of new polyclonal antibodies specific for 40 and 42 amino acid-long amyloid beta peptides: their use to examine the cell biology of presenilins and the immunohistochemistry of sporadic Alzheimer’s disease and cerebral amyloid angiopathy cases. Mol Med 1997; 3 : 695–707. [Google Scholar]
- Probst A, Basler V, Bron B, Ulrich J. Neuritic plaques in senile dementia of the Alzheimer type : a Golgi analysis in the hippocampal region. Brain Res 1983; 268 : 249–54. [Google Scholar]
- He Y, Delaère P, Duyckaerts C, Wasowicz M, Piette F, Hauw JJ. Two distinct ubiquitin immunoreactive senile plaques in Alzheimer’s disease: relationship with the intellectual status in 29 cases. Acta Neuropathol (Berl) 1993; 86 : 109–16. [Google Scholar]
- Eikelenboom P, Rozemuller JM, VanMuiswinkel FL. Inflammation and Alzheimer’s disease: Relationships between pathogenic mechanisms and clinical expression. Exp Neurol 1998; 154 : 89–98. [Google Scholar]
- Grignon Y, Duyckaerts C, Bennecib M, Hauw JJ. Cytoarchitectonic alterations in the supramarginal gyrus of late onset Alzheimer’s disease. Acta Neuropathol (Berl) 1998; 95 : 395–406. [Google Scholar]
- Gomez-Isla T, Price JL, McKeel DW, Morris JC, Growdon JH, Hyman BT. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J Neurosci 1996; 16 : 4491–500. [Google Scholar]
- Cras P, Smith MA, Richey PL, Siedlak SL, Mulvihill P, Perry G. Extracellular neurofibrillary tangles reflect neuronal loss and provide further evidence of extensive protein cross-linking in Alzheimer disease. Acta Neuropathol (Berl) 1995; 89 : 291–5. [Google Scholar]
- Stadelmann C, Deckwerth TL, Srinivasan A, et al. Activation of caspase-3 in single neurons and autophagic granules of granulovacuolar degeneration in Alzheimer’s disease. Evidence for apoptotic cell death. Am J Pathol 1999; 155 : 1459–66. [Google Scholar]
- Scheff SW, Price DA. Synapse loss in the temporal lobe in Alzheimer’s disease. Ann Neurol 1993; 33 : 190–9. [Google Scholar]
- Shimohama S, Kamiya S, Taniguchi T, Akagawa K, Kimura J. Differential involvement of synaptic vesicle and presynaptic plasma membrane proteins in Alzheimer’s disease. Biochem Biophys Res Commun 1997; 236 : 239–42. [Google Scholar]
- Terry RD, Masliah E, Salmon DP, et al. Physical basis of cognitive alterations in Alzheimer’s disease : synapse loss is the major correlate of cognitive impairment. Ann Neurol 1991; 30 : 572–80. [Google Scholar]
- Dickson DW, Crystal HA, Bevona C, Honer W, Vincent I, Davies P. Correlations of synaptic and pathological markers with cognition of the elderly. Neurobiol Aging 1995; 16 : 285–304. [Google Scholar]
- Delaère P, Duyckaerts C, Masters C, Piette F, Hauw JJ. Large amounts of neocortical ßA4 deposits without Alzheimer changes in a nondemented case. Neurosci Lett 1990; 116 : 87–93. [Google Scholar]
- Morris JC, Storandt M, McKeel DW Jr, et al. Cerebral amyloid deposition and diffuse plaques in « normal » aging: evidence for presymptomatic and very mild Alzheimer’s disease. Neurology 1996; 46 : 707–19. [Google Scholar]
- Hardy J. An ’anatomical cascade hypothesis’ for Alzheimer’s disease. Trends Neurosci 1992; 15 : 200–1. [Google Scholar]
- Duyckaerts C, Hauw JJ. Prevalence, incidence and duration of Braak’s stages in the general population: can we know ? Neurobiol Aging 1997; 18 : 362–9. [Google Scholar]
- Braak H, Braak E. Evolution of neuronal changes in the course of Alzheimer’s disease. In : Jellinger K, Fazekas F, Windisch M, eds. Ageing and dementia. Wien : Springer Verlag, 1998 : 127–40. [Google Scholar]
- Delacourte A, David JP, Sergeant N, et al. The biochemical pathway of neurofibrillary degeneration in aging and Alzheimer’s disease. Neurology 1999; 52 : 1158–65. [Google Scholar]
- Christie RH, Bacskai BJ, Zipfel WR, et al. Growth arrest of individual senile plaques in a model of Alzheimer’s disease observed by in vivo multiphoton microscopy. J Neurosci 2001; 21 : 858–64. [Google Scholar]
- Benveniste H, Einstein G, Kim KR, Hulette C, Johnson GA. Detection of neuritic plaques in Alzheimer’s disease by magnetic resonance microscopy. Proc Natl Acad Sci USA 1999; 96 : 14079–84. [Google Scholar]
- Dhenain M, Privat N, Duyckaerts C, Jacobs RE. Senile plaques do not induce susceptibility effects in T2*-weighted MR microscopic images. NMR Biomed 2002; 15 : 197–203. [Google Scholar]
- Zaim Wadghiri Y, Sigurdsson E, Tang C, et al. MR micro-imaging of contrast tagged amyloid plaques in transgenic mouse models of Alzheimer’s disease. In : 10th Scientific Meeting of the International Society for Magnetic Resonance in Medicine. Honolulu, USA: International Society for Magnetic Resonance in Medicine, 2002. [Google Scholar]
- Lee HJ, Zhang Y, Zhu C, Duff K, Pardridge WM. Imaging brain amyloid of Alzheimer disease in vivo in transgenic mice with an Aβ peptide radiopharmaceutical. J Cerebr Blood Flow Metab 2002; 22 : 223–31. [Google Scholar]
- Esiri MM, Hyman BT, Beyreuther K, Masters CI. Ageing and dementia. In : Graham DI, Lantos P, eds. Greenfield’s neuropathology. New York: Arnold, 1997 : 153–234. [Google Scholar]
- Najlerahim A, Bowen DM. Regional weight loss in the cerebral cortex and some subcortical nuclei in senile dementia of the Alzheimer type. Acta Neuropathol (Berl) 1989; 75 : 509–12. [Google Scholar]
- Duyckaerts C, Hauw JJ, Piette F, et al. Cortical atrophy in senile dementia of the Alzheimer type is mainly due to a decrease in cortical length. Acta Neuropathol (Berl) 1985; 66 : 72–4. [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease : report of the NINCDS-ADRDA work group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s disease. Neurology 1984; 19 : 939–44. [Google Scholar]
- Hubbard BM, Anderson JM. A quantitative study of cerebral atrophy in old age and senile dementia. J Neurol Sci 1981; 50 : 135–45. [Google Scholar]
- Erkinjuntti T, Lee DH, Gao F, et al. Temporal lobe atrophy on magnetic resonance imaging in the diagnosis of early Alzeimer’s disease. Arch Neurol 1993; 50 : 305–10. [Google Scholar]
- Killiany RJ, Gomez-Isla T, Moss M, et al. Use of structural magnetic resonance imaging to predict who will get Alzheimer’s disease. Ann Neurol 2000; 47 : 430–9. [Google Scholar]
- Deweer B, Lehericy S, Pillon B, et al. Memory disorders in probable Alzheimer’s disease: the role of hippocampal atrophy as shown with MRI. J Neurol Neurosurg Psychiatry 1995; 58 : 590–7. [Google Scholar]
- Barber R, Gholkar A, Scheltens P, Ballard C, McKeith IG, O’Brien JT. Medial temporal lobe atrophy on MRI in dementia with Lewy bodies: a comparison with Alzheimer’s disease, vascular dementia and normal ageing. Neurology 1999; 52 : 1153–8. [Google Scholar]
- Lehericy S, Baulac M, Chiras J, et al. Amygdalohippocampal MR volume measurements in the early stages of Alzheimer disease. Am J Neuroradiol 1994; 15 : 929–37. [Google Scholar]
- Fox NC, Crum WR, Scahill RI, Stevens JM, Janssen JC, Rossor MN. Imaging of onset and progression of Alzheimer’s disease with voxel-compression mapping of serial magnetic resonance images. Lancet 2001; 358 : 201–5. [Google Scholar]
- Yamauchi H, Fukuyama H, Nagahama Y, et al. Comparison of the pattern of atrophy of the corpus callosum in frontotemporal dementia, progressive supranuclear palsy, and Alzheimer’s disease. J Neurol Neurosurg Psychiatry 2000; 69 : 623–9. [Google Scholar]
- Hanyu H, Asano T, Sakurai H, et al. Diffusionweighted and magnetization transfer imaging of the corpus callosum in Alzheimer’s disease. J Neurol Sci 1999; 167 : 37–44. [Google Scholar]
- Kantarci K, Jack CR, Jr., Xu YC, et al. Mild cognitive impairment and Alzheimer disease: regional diffusivity of water. Radiology 2001; 219 : 101–7. [Google Scholar]
- Shonk T, Moats RA, Gifford P, et al. Probable Alzheimer’s disease. Diagnosis with proton MR spectroscopy. Radiology 1995; 185 : 65–72. [Google Scholar]
- Pettegrew JW, Klunk WE, Panchalingam K, McClure RJ, Stanley JA. Magnetic resonance spectroscopic changes in Alzheimer’s disease. Ann NY Acad Sci 1997; 826 : 282–306. [Google Scholar]
- Petit-Taboue MC, Landeau B, Desson JF, Desgranges B, Baron JC. Effects of healthy aging on the regional cerebral metabolic rate of glucose assessed with statistical parametric mapping. Neuroimage 1998; 7 : 176–84. [Google Scholar]
- Ashford JW, Shih WJ, Coupal J, et al. Single SPECT measures of cerebral cortical perfusion reflect time-index estimation of dementia severity in Alzheimer’s disease. J Nucl Med 2000; 41 : 57–64. [Google Scholar]
- Pasquier F, Lavenu I, Lebert F, Jacob B, Steinling M, Petit H. The use of SPECT in a multidisciplinary memory clinic. Dementia Geriatr Cogn Disord 1997; 8 : 85–91. [Google Scholar]
- Jelic V, Nordberg A. Early diagnosis of Alzheimer disease with positron emission tomography. Alzh Dis Ass Disord 2000; 14 (suppl 1) : S109–13. [Google Scholar]
- Grady CL. Brain imaging and age-related changes in cognition. Exp Gerontol 1998; 33 : 661–73. [Google Scholar]
- Rapoport SI. Functional brain imaging in the resting state and during activation in Alzheimer’s disease. Implications for disease mechanisms involving oxidative phosphorylation. Ann NY Acad Sci 1999; 893 : 138–53. [Google Scholar]
- Smith CD, Andersen AH, Kryscio RJ, et al. Altered brain activation in cognitively intact individuals at high risk for Alzheimer’s disease. Neurology 1999; 53 : 1391–6. [Google Scholar]
- Lehéricy S, Hirsch EC, Cervera-Piérot P, et al. Heterogeneity and selectivity of the degeneration of cholinergic neurons in the basal forebrain of patients with Alzheimer’s disease. J Comp Neurol 1993; 330 : 15–31. [Google Scholar]
- Horvath TB, Davis KL. Central nervous system disorders in aging. In : Schneider EL, Rowe JW, eds. Handbook of the biology of aging, 3rd ed. London : Academic Press, 1990 : 306–29. [Google Scholar]
- Bierer L, Haroutunian V, Gabriel S, et al. Neurochemical correlates of dementia severity in Alzheimer’s disease: Relative importance of the cholinergic deficits. J Neurochem 1995; 64 : 749–60. [Google Scholar]
- Volkow ND, Ding YS, Fowler JS, Gatley SJ. Imaging brain cholinergic activity with positron emission tomography: its role in the evaluation of cholinergic treatments in Alzheimer’s dementia. Biol Psychiatry 2001; 49 : 211–20. [Google Scholar]
- Meltzer CC, Smith G, DeKosky ST, et al. Serotonin in aging, late-life depression, and Alzheimer’s disease: The emerging role of functional imaging. Neuropsychopharmacology 1998; 18 : 407–30. [Google Scholar]
- Duyckaerts C, Bennecib M, Grignon Y, et al. Modeling the relation between neurofibrillary tangles and intellectual status. Neurobiol Aging 1997; 18 : 267–73. [Google Scholar]
Liste des tableaux
Techniques d’imagerie cérébrale permettant de réaliser des images fonctionnelles. TEP : tomographie par émission de positons. TEMP : tomographie par émission de photon unique. IRM : imagerie par résonance magnétique.
Liste des figures
|
Figure 1. Aspect des lésions observées dans la maladie d’Alzheimer à l’examen microscopique. Toutes les photographies ont été prises avec un objectif x100 ; le trait de l’image A représente 20 µm. A. Plaque sénile ; immunohistochimie du peptide Aβ. Un dépôt dense de peptide Aβ (grande flèche) occupe le centre de la plaque sénile. Elle est entourée d’une couronne claire (correspondant aux prolongements nerveux de l’image C), puis d’un halo de dépôt diffus (petites flèches). Un noyau de macrophage est marqué par une tête de flèche. B. Plaque sénile colorée par le rouge Congo, colorant qui se fixe sélectivement sur les substances amyloïdes. La partie amyloïde de la plaque sénile (correspondant au centre dense de la photo A) est indiquée par une flèche. La tête de flèche marque le noyau d’un macrophage. C. Plaque sénile ; immunohistochimie de la protéine tau. Le centre de la plaque (grande flèche) est à peine visible. Il est entouré de prolongements nerveux dégénérés, marqués par l’anticorps anti-tau (petites flèches). Le noyau de deux macrophages est indiqué par des têtes de flèches. D. Dégénérescence neurofibrillaire ; immunohistochimie de la protéine tau. Deux neurones sont visibles (marqués a et b). Le noyau est indiqué par une grosse tête de flèche, le nucléole par une petite tête de flèche. La dégénérescence neurofibrillaire est constituée par des fibrilles de protéine tau dans le corps cellulaire du neurone a (flèches). |
| Dans le texte | |
|
Figure 2. Représentation schématique de la progression de la pathologie neurofibrillaire dans le cortex cérébral. A. Vue latérale. B. vue médiale. La première aire corticale à être affectée par les dégénérescences neurofibrillaires est le cortex entorhinal (marqué 1). A l’étape suivante, les lésions sont observées dans l’hippocampe (flèche 2) et le cortex rétrosplénial. La pathologie neurofibrillaire intéresse ensuite la partie antérieure du lobe temporal (flèche 3). Les aires associatives uni- puis multimodales frontale (AMAfr) et temporo-pariéto-occipitales (AMAtpo) sont concernées à l’étape suivante (flèches marquées 4). Les aires primaires (motrice = M1, somesthésique = S1, auditive = A1, visuelle = V1) sont atteintes en dernier lieu (flèches 5). (D’après [24, 25, 59]). |
| Dans le texte | |
|
Figure 3. Image obtenue par l’analyse en microscopie-IRM d’échantillon de tissus d’un patient atteint de maladie d’Alzheimer. Cette image, pondérée en T2*, a une résolution isotrope de 23,4 µm par voxel. Les structures punctiformes hypo-intenses (flèche rouge épaisse) ont été interprétées comme des plaques séniles [27]. Une étude plus récente suggère qu’il s’agirait de sections de vaisseaux, tout comme les structures plus allongées (flèches rouges fines) [28]. |
| Dans le texte | |
|
Figure 4. Anatomie hippocampique par IRM. Coupes coronales obliques perpendiculaires au grand axe des formations hippocampiques passant par l’uncus (A), le corps (B) et la queue (C) de l’hippocampe. Les formations hippocampiques sont indiquées par les flèches rouges, l’amygdale par la flèche jaune. |
| Dans le texte | |
|
Figure 5. Atrophie hippocampique dans la maladie d’Alzheimer. Coupe coronale pondérée en T1 passant par le corps des formations hippocampiques (flèches) d’un sujet témoin ( A ) et d’un patient atteint de maladie d’Alzheimer ( B ). Il existe une atrophie importante des formations hippocampiques du patient atteint de maladie d’Alzheimer. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.