")
")
| Issue |
Med Sci (Paris)
Volume 18, Number 1, Janvier 2002
|
|
|---|---|---|
| Page(s) | 19 - 22 | |
| Section | Le Magazine : Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/200218119 | |
| Published online | 15 January 2002 | |
La thrombine et ses récepteurs : implications dans l’hémostase et le développement embryonnaire
Thrombin and its recep-tors: beyond hemosta-sis, a rôle in embryonic development
E9907 Inserm, Faculté Xavier Bichat, BP 416, 16, rue Henri Huchard, 75870 Paris Cedex 18, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
La thrombine est le produit d’une cascade finement réglée d’activation de zymogènes déclenchée par la rencontre des facteurs plasmatiques de la coaguation et du facteur tissulaire exposé auniveau d’une lésion vasculaire. La thrombine clive le fibrinogène soluble en monomères qui forment par polymérisation le réseau de fibrine insoluble. Ce processus associé à l’activation de plaquettes au contact de la matrice sous-endothéliale aboutit à la formation du caillot assurant l’hémostase. La production inappropriée de thrombine et /ou l’activation intempestive des plaquettes sont le fondement des événements thrombo-emboliques. La régulation de l’activité de la thrombine et de sa formation est donc cruciale. La thrombine a la capacité de contrôler positivement et négativement sa propre formation en activant d’une part les cofacteurs V et VIII et d’autre part la protéine C, ellemême responsable de l’inactivation de ces cofacteurs. Parmi les enzymes de la coagulation, la thrombine se caractérise par une activité cellulaire importante. En effet, elle est un puissant activateur des plaquettes et de multiples études ont montré qu’elle induit des signaux dans la plupart des cellules étudiées in vitro.
Après un quart de siècle de recherches infructueuses, un récepteur de la thrombine a été simultanément identifié et son gène cloné par différents groupes [1, 2]. Ce récepteur appartient à la superfamille des récepteurs à sept domaines transmembranaires couplés aux protéines G mais possède un mécanisme d’action particulier. La thrombine se lie au récepteur par un site spécifique et le clive, démasquant une nouvelle extrémité N-terminale qui agit comme peptide ago-niste interne (Figure 1). Du fait de son clivage, ce récepteur appelé PAR1 (protease activated recep-tor-1) est à usage unique. PAR1 est le premier d’une famille de quatre comportant PAR3 et PAR4 également récepteurs de la thrombine [3], PAR2 n’étant en revanche pas activé par la thrombine. La distribution tissulaire et cellulaire large de PAR1 rend compte des effets multiples de la thrombine observés in vitro.
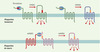 |
Figure 1. Les PAR des plaquettes humaines et de souris. Les plaquettes humaines expriment PAR1 et PAR4 qui peuvent être indépendamment activés par la thrombine (en bleu). La liaison de la thrombine à PAR1 au niveau d’un site complémentaire permet le clivage du site peptidique cible, la nouvelle extrémité N-terminale démasquée agissant en tant que peptide agoniste interne. PAR4 ne possédant pas de site complémentaire de liaison de la thrombine, n’est activé qu’à forte concentration d’enzyme. Les plaquettes de souris expriment PAR3 et PAR4. PAR3 est incapable d’induire des signaux par lui-même, mais présente un site de liaison de la thrombine et agit comme cofacteur du clivage et de l’activation de PAR4. |
Cependant, in vivo, le rôle de la thrombine au niveau cellulaire n’est pas clair mais l’obtention de souris déficientes en protéines impliquées dans la formation de la thrombine ou de son récepteur apporte de nouveaux éléments.
L’invalidation du gène de PAR1 [4] a révélé deux notions importantes : d’une part, l’activation des plaquettes par la thrombine implique d’autres récepteurs que PAR1, et d’autre part le rôle physiologique de la thrombine et de l’activation de son récepteur dépasse le strict cadre de l’hémostase, puisqu’il contribue au développement embryonnaire. Shaun Coughlin et son groupe ont très largement contribué à l’identification des récepteurs de la thrombine et eurs deux derniers articles [5, 6] vont plus loin dans la description de leur rôle tant au plan de l’hémostase que de l’embryogenèse.
Rôle des récepteurs de la thrombine dans l’hémostase et la thrombose
L’activation des plaquettes par la thrombine fait intervenir au moins deux PAR (Figure 1) de manière sensiblement différente chez l’homme et la souris. Les plaquettes humaines expriment PAR1 et PAR4, les deux récepteurs étant activables indépendamment l’un de l’autre. PAR1 est activé par la thrombine à faible concentration alors que, en raison de sa plus faible affinité pour l’enzyme, PAR4 est activé à plus forte concentration [3]. En revanche, les plaquettes de souris expriment PAR3 et PAR4, tous les deux nécessaires à l’activation des plaquettes. L’invalidation des gènes de PAR3 [7] et de PAR4 [6] a montré que PAR3 est insuffisant pour induire l’activation des plaquettes mais que en liant la thrombine, il se comporte comme cofacteur de l’activation de PAR4.
L’invalidation du gène de PAR4 a pour conséquence un allongement très important du temps de saignement confirmant que l’activation des plaquettes par la thrombine est nécessaire pour assurer l’hémostase. Par ailleurs, dans un modèle in vivo de thrombose mésentérique, le temps d’occlusion artériolaire et le nombre d’artérioles occluses sont réduits dans le cas des souris déficientes en PAR4 indiquant que l’activation des plaquettes par la thrombine joue un rôle crucial dans ce modèle de thrombose.
L’observation que l’ablation de la signalisation induite par la thrombine dans la plaquette de souris protège contre la thrombose et que des anticorps anti-PAR réduisent l’occlusion cyclique des vaisseaux dans un autre modèle animal de thrombose, suggère que l’inhibition des signaux plaquettaires induits par la thrombine pourrait constituer une stratégie de prévention et de traitement des thromboses. Cependant quelques réserves peuvent être émises quant à la faisabilité et à l’innocuité d’une telle approche. En effet, en raison du mécanisme spécifique d’activation des plaquettes humaines par la thrombine, il serait nécessaire de bloquer simultanément l’activation de PAR1 et de PAR4 pour être efficace. Il n’est de plus pas formellement exclu que d’autres récepteurs, en particulier le complexe des glycoprotéines Ib-IX-V, soient également impliqués dans l’activation des plaquettes par la thrombine [8]. De plus, le temps de saignement très prolongé des souris PAR4−/− ferait craindre un risque hémorragique élevé.
Rôle des récepteurs de la thrombine dans le développement
L’analyse croisée des résultats de différentes invalidations de gènes touchant soit les facteurs de la coagulation soit les PAR indique clairement l’implication de la cascade de la coagulation et des PARs dans le développement vascuaire chez l’embryon.
Le nombre de souriceaux PAR4−/− et PAR3−/− obtenus par croisement de souris hétérozygotes pour le déficit respecte la fréquence mendélienne et ces souris se développent normalement [6, 7]. En revanche, 50% des embryons PAR1−/−meurent prématurément d’hémorragie et d’insuffisance cardiovasculaire [4, 5]. Des hémorragies microscopiques sont observées dès J8,5 et à J9,5 les hémorragies sont macroscopiques avec présence de sang dans les cavités péricardique, amniotique et péritonéale [5].
Différents arguments indiquent que ces hémorragies ne sont pas dues à un défaut de l’hémostase : les plaquettes ne sont pas encore produites à ce stade du développement et lorsqu’elles sont produites, elles sont normalement activées par la thrombine via PAR3/PAR4. En revanche des anomalies vasculaires seraient à l’origine des hémorragies. Un retard de développement vasculaire, des brèches dans la paroi du sinus veineux et des anomalies des vaisseaux du sac vitellin sont observés. En raison de l’expression endothéliale de PAR1, les auteurs [5] ont émis l’hypothèse que la perte des signaux couplés à PAR1 dans les cellules endothéliales et de l’endocarde serait à l’origine des anomalies vasculaires. De fait, dans les animaux PAR1−/−, la réintroduction de PAR-1 dans es cellules endothéliales par un transgène dont l’expression est sous le contrôle du promoteur/en/iancer Tie-2 qui cible spécifiquement les cellules endothéliales, réduit de trois à quatre fois la mortalité embryonnaire.
Les signaux couplés à PAR1 semblent donc cruciaux pour le dévelopement vasculaire. Mais la thrombine est-elle alors l’activateur endogène de PAR1 et dans quelle mesure les protéines de la coagulation agissant en amont de la thrombine sont-elles impliquées dans ce processus (Figure 2) ? Le phénotype des souris déficientes en prothrombine (ou facteur II) (F11−/−) est proche de celui de souris PAR1−/− avec une vague de mortalité précoce ente J9,5 et J11,5 [9] et des hémorragies [9]. De même, le déficit en Facteur V (FV), cofacteur de l’activation de la prothrombine par le Facteur Xa (FXa) résulte dans le décès prématuré (J9,5-J10,5) de ~50% des embryons [10]. Ces observations sont en faveur d’un rôle précoce de la thrombine dans le développement vasculaire. Il est à noter que les premiers signes d’anomalie du développement vasculaire des souris Flh−/− et PAR1−/− précèdent la formation du bourgeon hépatique (J10,5), lieu de synthèse de la prothrombine, suggérant une expression embryonnaire extra-hépatique. Concernant les autres facteurs de la coagulation en amont de l’activation de la prothrombine, le déficit en FX résulte aussi en une létalité embryonnaire partielle et plus tardive (J12,5), liée à des hémorragies sans modifications vasculaires [11] alors que le déficit en facteur VII n’a pas de conséquences sur le développement embryonnaire [12]. En revanche l’invalidation du gène du facteur tissulaire (FT)1 est létale, avec mort de tous les embryons FT−/− entre J9,5 et J11,5 par fuite des globules rouges hors des vaisseaux embryonnaires et extra-embryonnaires [13], indiquant que le FT est indispensable pour l’établissement et/ou le maintien de l’intégrité vascuaire au cours du développement embryonnaire. Griffin et al. [5] ont observé un phénotype et un degré de mortalité similaire pour les embryons PAR1−/−FV−/−. La comparaison du phénotype de ces doubles knock-out (100% de mortalité) et du phénotype qu’entraîne ’invalidation d’un seul gène (50% de mortalité) suggère que PAR1 peut être activé en l’absence de FV, soit par les traces de thrombine qui peuvent être produites par le FXa, en l’absence de FVa, soit par d’autres protéases. Il a ainsi été récemment montré que PAR1 pouvait être efficacement activé par le complexe FT-FVIIa-FX [14]. Par ailleurs, d’autres PAR que PAR1 peuvent être impliqués dans le développement vascuaire. En effet, les cellules endothéliales expriment PAR2 qui peut lui-même être activé par le FXa couplé à son récepteur EPR-1 (effector-protease receptor-1) et le déficit en PAR2 s’accompagne d’une surmortalité pré- et postnatale [15].
 |
Figure 2. Modèle d’action de la thrombine, de ses récepteurs et des protéines de coagulation d’amont dans l’hémostase et le développement. La voie classique d’activation de la prothrombine est représentée. Les protéines dont le déficit est associé à des anomalies du développement vasculaire sont indiquées en rouge. |
Chez l’homme le déficit héréditaire en FV ou para-hémophilie est rare (1/106), la survie des patients étant imputée à la production de traces de FV. Un déficit complet en prothrombine et en FX est rarement rencontré.
Concernant PAR1 et le FT, il n’a jusqu’à présent été rapporté aucun cas de déficit, suggérant que de tels déficits sont létaux.
L’ensemble de ces investigations permet une meilleure compréhension des mécanismes de l’hémostase et l’ouverture de nouvelles voies dans la prévention et le traitement des thromboses.
Par ailleurs, elles mettent en évidence un rôle original du système de la coagulation dont certaines protéines sont adaptées pour contribuer à la croissance et à la stabilité vasculaire au cours du développement embryonnaire.
Le FT est une protéine membranaire qui agit comme cofacteur du facteur VIIa et dont l’expression tissulaire normale est physiquement bien séparée du flux sanguin. L’exposition du FT par une ésion vasculaire est considérée comme élément déclenchant de la coagulation.
Références
- Vu TK, Hung DT, Wheaton VI, Coughlin SR. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cetl 1991; 64 : 1057–68. [Google Scholar]
- Rasmussen UB, Vouret-Craviari V, Jallat S, et al. cDNA cloning and expression of a hamster alpha-thrombin receptor coupled to Ca2+ mobilization. FEBS Lett, 1991; 288 : 123–8. [Google Scholar]
- Coughlin SR. Thrombin signalling and protease activated receptors. Nature 2000; 407: 258–64. [Google Scholar]
- Connolly AJIshihara H, Kahn ML, Farese RV, Coughlin SR. Role of the thrombin receptor in development and evidence for a second receptor. Nature 1996; 381 : 516–9. [Google Scholar]
- Griffin CT, Srinivasan Y, Zheng YW, Huang W, Coughlin SR. A role for thrombin receptor signaling in endothelial cells during embryonic development. Science 2001; 293 : 1666–70. [Google Scholar]
- Sambrano GR, Weiss EJ, Zheng YW, Huang W, Coughlin SR. Role of thrombin signaling in platelets in haemostasis and thrombosis. Nature 2001; 413 : 74–8. [Google Scholar]
- Kahn ML, Zheng YW, Huang W, et al. A dual thrombin receptor system for platelet activation. Nature 1998; 394 : 690–4. [Google Scholar]
- Berndt MC, Shen Y, Dopheide SM, Gardiner EE, Andrews RK. The vascular biology of the glycoprotein Ib-IX-V complex. Thromb Haemost 2001; 86 : 178–88. [Google Scholar]
- Sun WY, Witte DP, Degen JL, et al. Prothrombin deficiency results in embryonic and neonatal lethality in mice. Proc Natl Acad Sci USA, 1998; 95 : 7597–602. [Google Scholar]
- Cui J, et al. Fatal haemorrhage and incomplete block to embryogenesis in mice lacking coagulation factor V. Nature 1996; 384 : 66–8. [Google Scholar]
- Dewerchin M, Liang Z, Moons L, et al. Blood coagulation factor X deficiency causes partial embryonic lethality and fatal neonatal bleeding in mice. Thromb Haemost 2000; 83 : 185–90. [Google Scholar]
- Rosen ED, Chan JC, Idusogie E, et al. Mice lacking factor VII develop normally but suffer fatal perinatal bleeding. Nature 1997; 390 : 290–4. [Google Scholar]
- Bugge TH, Xiao Q, Kombrinck KW, et al. Fatal embryonic bleeding events in mice lacking tissue factor, the cell- associated initiator of blood coagulation. Proc Natl Acad Sci USA 1996; 93 : 6258–63. [Google Scholar]
- Riewald M, Ruf W. Mechanistic coupling of protease signaling and initiation of coagulation by tissue factor. Proc Natl Acad Sci USA 2001; 98 : 7742–7. [Google Scholar]
- Damiano BP, Cheung WM, Santulli RJ, et al. Cardiovascular responses mediated by protease-activated receptor-2 (PAR- 2) and thrombin receptor (PAR-1) are distinguished in mice deficient in PAR-2 or PAR-1. j Pharmacol Exp Ther 1999; 288 : 671–8. [Google Scholar]
© 2002 médecine/sciences - Inserm / SRMS
Liste des figures
|
Figure 1. Les PAR des plaquettes humaines et de souris. Les plaquettes humaines expriment PAR1 et PAR4 qui peuvent être indépendamment activés par la thrombine (en bleu). La liaison de la thrombine à PAR1 au niveau d’un site complémentaire permet le clivage du site peptidique cible, la nouvelle extrémité N-terminale démasquée agissant en tant que peptide agoniste interne. PAR4 ne possédant pas de site complémentaire de liaison de la thrombine, n’est activé qu’à forte concentration d’enzyme. Les plaquettes de souris expriment PAR3 et PAR4. PAR3 est incapable d’induire des signaux par lui-même, mais présente un site de liaison de la thrombine et agit comme cofacteur du clivage et de l’activation de PAR4. |
| Dans le texte | |
|
Figure 2. Modèle d’action de la thrombine, de ses récepteurs et des protéines de coagulation d’amont dans l’hémostase et le développement. La voie classique d’activation de la prothrombine est représentée. Les protéines dont le déficit est associé à des anomalies du développement vasculaire sont indiquées en rouge. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.