")
")
| Issue |
Med Sci (Paris)
Volume 28, Number 6-7, Juin–Juillet 2012
|
|
|---|---|---|
| Page(s) | 580 - 582 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/2012286008 | |
| Published online | 16 July 2012 | |
Faut-il bloquer l’interleukine-1 dans l’athérothrombose ?
Should we block interleukin-1 in atherothrombosis?
Université Paris Diderot, Sorbonne Paris-Cité, Inserm UMRS 698, CHU Xavier Bichat, 16, rue Henri Huchard, 75018 Paris, France
Photo : structure monomérique de l’IL-1β (© EMBL-EBI).
En dépit d’importants investissements et des stratégies thérapeutiques disponibles - prévention des facteurs de risque et traitements interventionnels -, les maladies athérothrombotiques restent une des premières causes de morbidité et mortalité dans les pays développés. Ce constat nous force à approfondir ce que sont réellement les lésions d’athérothrombose chez l’homme, à déterminer comment elles se développent et comment elles évoluent vers leur expression clinique.
Interleukine-1 et lésions d’athérothrombose
Depuis plus de dix ans, le paradigme dominant est que l’inflammation est le moteur de la progression des lésions d’athérothrombose [1], alors même que les traitements anti-inflammatoires classiques augmentent la fréquence de l’expression clinique de la maladie. Dans ce contexte du « tout inflammatoire », l’intérêt s’est récemment porté sur l’interleukine 1 bêta (IL-1β), une cytokine pro-inflammatoire produite par les plaques à tous les stades d’athérome, en rapport avec la surcharge lipidique [2] et la cristallisation du cholestérol [3]. Contrairement à l’IL-1α dont elle partage les activités biologiques, l’IL-1β est une cytokine sécrétée après son clivage par la caspase 1, cette protéase étant elle-même activée par l’« inflammasome ». Si les sources principales d’IL-1β sont les leucocytes, les plaquettes et de très nombreuses cellules stromales sont aussi capables de la produire. Les récepteurs de l’IL-1 sont également présents sur de nombreuses cellules de l’organisme, et pas seulement sur les leucocytes. L’IL-1β et son récepteur principal, l’IL-1RI, représentent donc un système extrêmement ubiquitaire.
Essai thérapeutique de blocage de l’interleukine-1 dans la maladie athéromateuse
Un essai clinique de blocage de l’IL-1β par un anticorps monoclonal humanisé, le Canakinumab (ILARIS®, étude CANTOS [Canakinumab anti-inflammatory thrombosis outcomes study]), est en cours dans l’athérothrombose [4], et ce malgré l’arrêt - pour une raison qui n’a pas été rendue publique - d’un essai du Canakinumab dans la polyarthrite rhumatoïde chez l’homme (clinicaltrials.gov, Identifier NCT00784628). Le Canakinumab a cependant été approuvé par la FDA (Food and drug administration) (États-Unis) et par l’EMA (European medicines agency) (Europe) dans l’indication des cryopyrinopathies de l’enfant, une pathologie s’accompagnant de mutations dans le gène NLRP3 (NLR family, pyrin domain containing 3) codant pour une protéine impliquée dans l’activation de la caspase 1. L’Anakinra, un antagoniste synthétique des récepteurs de l’IL-1β, est peu efficace dans la polyarthrite rhumatoïde [5] et n’a pas encore été testé dans l’athérothrombose humaine. La souris apoE-/-, lorsqu’elle est soumise à un régime hypercholestérolémique, développe une hypertension artérielle pulmonaire (HTAP) caractérisée par une prolifération diffuse des cellules musculaires lisses (CML). Ces souris, lorsqu’elles sont croisées avec d’autres souris invalidées pour l’IL-1RI, ou lorsqu’elles reçoivent de l’Anakinra, ne développent pas ce phénotype d’HTAP [6]. De même, dans un modèle de prolifération intimale des CML chez la souris, l’invalidation de l’IL-1RI prévient cette prolifération [7]. Ces résultats montrent que l’IL-1β pourrait être impliquée dans la prolifération des CML et l’acquisition d’un phénotype cicatrisant et sécrétoire de matrice extracellulaire.
De façon inattendue, le groupe de Gary K. Owens [8] vient de montrer que l’inactivation génétique de l’IL-1β chez la souris apoE-/- conduit à une réduction sévère (60 %) de la lumière artérielle et ce malgré une réduction de la surface des plaques lipidiques (d’environ 30 %). Ceci s’explique par l’abolition, chez ces souris, du remodelage excentrique de la média pariétale en regard de la plaque, un processus de compensation qui permet le maintien du diamètre de la lumière artérielle malgré la croissance de la lésion athéromateuse. L’invalidation de l’IL-1RI s’accompagne également d’une diminution de la taille de la chape fibreuse et cellulaire et de l’augmentation de la densité de globules rouges dans les plaques. Ces données suggèrent qu’en l’absence de signalisation de l’IL-1β, la maladie athéromateuse évolue défavorablement avec des sténoses plus sévères et des plaques plus vulnérables (Figure 1). Cette étude est donc un signal d’alarme sur les risques d’un blocage pharmacologique de la voie de l’IL-1.
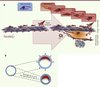 |
Figure 1. Mécanismes de formation de la plaque d’athérome chez l’homme. A. L’athérome humain est déclenché par la rétention intimale de cholestérol. Cette lésion intimale, ou strie lipidique, est constituée également par des CML produisant des protéoglycanes polyanioniques interagissant avec les acides aminés chargés de l’apoB (apolipoprotéine B) des LDL (low density lipoprotein). La rétention des LDL donne alors lieu à la formation de cellules spumeuses, chargées de gouttelettes lipidiques et de cristaux de cholestérol. La plaque initiale, ou lésion fibrolipidique, est caractérisée par la formation d’un coeur lipidique extracellulaire et la mort des CML spumeuses, qui seront recouvertes ultérieurement par une chape fibrocellulaire sécrétée par d’autres CML, plus résistantes à l’agression lipidiques [9]. En réponse à l’accumulation intimale de lipides, les CML de la média orchestrent la réponse angiogénique depuis l’adventice vers la plaque [12]. La néoangiogenèse intraplaque est l’étape cruciale signant l’évolution de la plaque vers un phénotype compliqué. La média artérielle est physiologiquement avasculaire. Elle est un site immun privilégié, peu ou pas accessible aux leucocytes circulants en l’absence de néovascularisation permettant leur extravasation. Immature et perméable, le néoréseau veinocapillaire transpariétal, où règne une pression propice à l’extravasation des leucocytes vers la plaque, sera le vecteur d’hémorragies intraplaques [13]. B. Le travail de Alexander et al. [8] montre que le blocage de la signalisation de l’IL-1 conduit à un remodelage sténosant et vulnérable des plaques (en rouge, accolées à la face interne de la paroi vasculaire en bleu). |
Athérogenèse et inflammation : un paradigme abusif ?
Ce travail offre l’opportunité de discuter plusieurs points essentiels concernant l’utilisation du terme inflammation dans la pathologie athéromateuse : existe-t-il une véritable relation de causalité entre l’extravasation leucocytaire dans les plaques et leur évolution morbide ? Quelles sont les limites des modèles murins dans la compréhension de la maladie humaine ?
Le processus d’athérogenèse chez l’homme, succinctement décrit dans la Figure 1, met en lumière les limites des modèles murins dans lesquels l’angiogenèse transpariétale et les hémorragies intraplaques sont extrêmement rares. Ce schéma place aussi les CML au centre des évolutions successives de la pathologie chez l’homme : prolifération intimale initiale, cellules spumeuses, chape fibrocellulaire, organisation des réponses adventitielles [9]. Si les CML peuvent adopter des phénotypes aussi divers que contractile, prolifératif, sécrétoire, phagocytaire, voire ostéoblastique, c’est qu’elles gardent une forte plasticité phénotypique chez l’adulte [9, 14] (→).
(→) Voir la Nouvelle de K. Treguer et al., page 584 de ce numéro
La cristallisation du cholestérol dans le microenvironnement des CML est un évènement macromoléculaire probablement capital dans ces changements fonctionnels, comme cela a été montré pour le macrophage [3]. Les capacités cicatrisantes des CML limitent le risque de rupture des plaques, et leur mort participe de la pathogenèse de plaques de plus en plus complexes. Les CML mériteraient donc d’occuper une place plus importante dans l’athérosclérose humaine. Or, l’implication préférentielle des leucocytes mononucléés, suggérée par les travaux chez la souris, nous incline à utiliser des stratégies anti-inflammatoires possiblement inappropriées car ne pointant pas les bonnes cibles et/ou sans doute trop radicales.
À cet égard, l’extravasation de monocytes et de lymphocytes vers la plaque est souvent présentée comme étant un processus délétère, alors que la réponse immune innée et certaines réponses adaptatives peuvent être athéroprotectrices [10, 11]. Dans un tel contexte de mixité et de plasticité des cellules stromales et des cellules d’origine hématopoïétique, l’impact du blocage d’une cytokine telle que l’IL-1β, produite par et agissant sur les deux familles de cellules, est difficile à prédire. C’est précisément ce que montre le travail d’Alexander et al. [8] dans lequel l’inhibition de la signalisation de l’IL-1β n’est pas univoque et intègre à la fois des effets bénéfiques sur les leucocytes et la taille des lésions, et des effets délétères sur le remodelage sténosant et vulnérable des plaques via les CML.
L’ensemble de la communication scientifique concernant l’athérothrombose pose finalement des questions d’ordre sémantique, épistémologique et éthique autour de l’emploi abusif du mot inflammation. L’emploi de ce terme, initialement issu de la séméiologie médicale, est, dans les sciences biologiques, dans la plupart des cas approximatif, ne traduisant pas de façon analytique le phénomène étudié. De quoi s’agit-il ? De signalisation intracellulaire ? De sécrétion de cytokines ou chémokines ? De phénomènes cellulaires tels que l’extravasation de leucocytes ? Si oui, quels phénomènes parmi ceux-ci et de quelles fonctions sont-ils dotés ? Le progrès médical ne peut se suffire de telles approximations, surtout, lorsque des essais thérapeutiques incluant la participation humaine sont en jeu.
Liens d’Intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Références
- Ross R. Atherosclerosis-an inflammatory disease. N Engl J Med 1999 ; 340 : 115–126. [Google Scholar]
- Clarke MC, Talib S, Figg NL, Bennett MR. Vascular smooth muscle cell apoptosis induces interleukin-1-directed inflammation: effects of hyperlipidemia-mediated inhibition of phagocytosis. Circ Res 2010 ; 106 : 363–372. [CrossRef] [PubMed] [Google Scholar]
- Duewell P, Kono H, Rayner KJ, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010 ; 464 : 1357–1361. [CrossRef] [PubMed] [Google Scholar]
- Ridker PM, Thuren T, Zalewski A, Libby P. Interleukin-1 beta inhibition and the prevention of recurrent cardiovascular events: rationale and design of the Canakinumab anti-inflammatory thrombosis outcomes study (CANTOS). Am Heart J 2011 ; 162 : 597–605. [CrossRef] [PubMed] [Google Scholar]
- Mertens M, Singh JA. Anakinra for rheumatoid arthritis: a systematic review. J Rheumatol 2009 ; 36 : 1118–1125. [CrossRef] [PubMed] [Google Scholar]
- Lawrie A, Hameed AG, Chamberlain J, et al. Paigen diet-fed apolipoprotein E knockout mice develop severe pulmonary hypertension in an interleukin-1-dependent manner. Am J Pathol 2011 ; 179 : 1693–1705. [CrossRef] [PubMed] [Google Scholar]
- Saxena A, Rauch U, Berg KE, et al. The vascular repair process after injury of the carotid artery is regulated by IL-1RI and MyD88 signalling. Cardiovasc Res 2011 ; 91 : 350–357. [CrossRef] [PubMed] [Google Scholar]
- Alexander MR, Moehle CW, Johnson JL, et al. Genetic inactivation of IL-1 signaling enhances atherosclerotic plaque instability and reduces outward vessel remodeling in advanced atherosclerosis in mice. J Clin Invest 2012 ; 122 : 70–79. [CrossRef] [PubMed] [Google Scholar]
- Lacolley P, Regnault V, Nicoletti A, et al. The vascular smooth muscle cell in arterial pathology: a cell that can take on multiple roles. Cardiovasc Res 2012 (sous-presse). [Google Scholar]
- Ait-Oufella H, Salomon BL, Potteaux S, et al. Natural regulatory T cells control the development of atherosclerosis in mice. Nat Med 2006 ; 12 : 178–180. [CrossRef] [PubMed] [Google Scholar]
- Caligiuri G, Nicoletti A, Poirier B, Hansson GK. Protective immunity against atherosclerosis carried by B cells of hypercholesterolemic mice. J Clin Invest 2002 ; 109 : 745–753. [CrossRef] [PubMed] [Google Scholar]
- Ho-Tin-Noé B, Le Dall J, Gomez D, et al. Early atheroma-derived agonists of peroxisome proliferator-activated receptor-γ trigger intramedial angiogenesis in a smooth muscle cell-dependent manner. Circ Res 2011 ; 109 : 1003–1014. [CrossRef] [PubMed] [Google Scholar]
- Le Dall J, Ho-Tin-Noé B, Louedec L, et al. Immaturity of microvessels in haemorrhagic plaques is associated with proteolytic degradation of angiogenic factors. Cardiovasc Res 2010 ; 85 : 184–193. [CrossRef] [PubMed] [Google Scholar]
- Tréguer K, Heydt S, Hergenreider E. Protection des vaisseaux sanguins contre l’athérosclérose : le rôle des miARN sécrétés par l’endothélium. Med Sci (Paris) 2012 ; 28 : 584–587. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
© 2012 médecine/sciences – Inserm / SRMS
Liste des figures
|
Figure 1. Mécanismes de formation de la plaque d’athérome chez l’homme. A. L’athérome humain est déclenché par la rétention intimale de cholestérol. Cette lésion intimale, ou strie lipidique, est constituée également par des CML produisant des protéoglycanes polyanioniques interagissant avec les acides aminés chargés de l’apoB (apolipoprotéine B) des LDL (low density lipoprotein). La rétention des LDL donne alors lieu à la formation de cellules spumeuses, chargées de gouttelettes lipidiques et de cristaux de cholestérol. La plaque initiale, ou lésion fibrolipidique, est caractérisée par la formation d’un coeur lipidique extracellulaire et la mort des CML spumeuses, qui seront recouvertes ultérieurement par une chape fibrocellulaire sécrétée par d’autres CML, plus résistantes à l’agression lipidiques [9]. En réponse à l’accumulation intimale de lipides, les CML de la média orchestrent la réponse angiogénique depuis l’adventice vers la plaque [12]. La néoangiogenèse intraplaque est l’étape cruciale signant l’évolution de la plaque vers un phénotype compliqué. La média artérielle est physiologiquement avasculaire. Elle est un site immun privilégié, peu ou pas accessible aux leucocytes circulants en l’absence de néovascularisation permettant leur extravasation. Immature et perméable, le néoréseau veinocapillaire transpariétal, où règne une pression propice à l’extravasation des leucocytes vers la plaque, sera le vecteur d’hémorragies intraplaques [13]. B. Le travail de Alexander et al. [8] montre que le blocage de la signalisation de l’IL-1 conduit à un remodelage sténosant et vulnérable des plaques (en rouge, accolées à la face interne de la paroi vasculaire en bleu). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.