")
")
Figure 2
Télécharger l'image originale
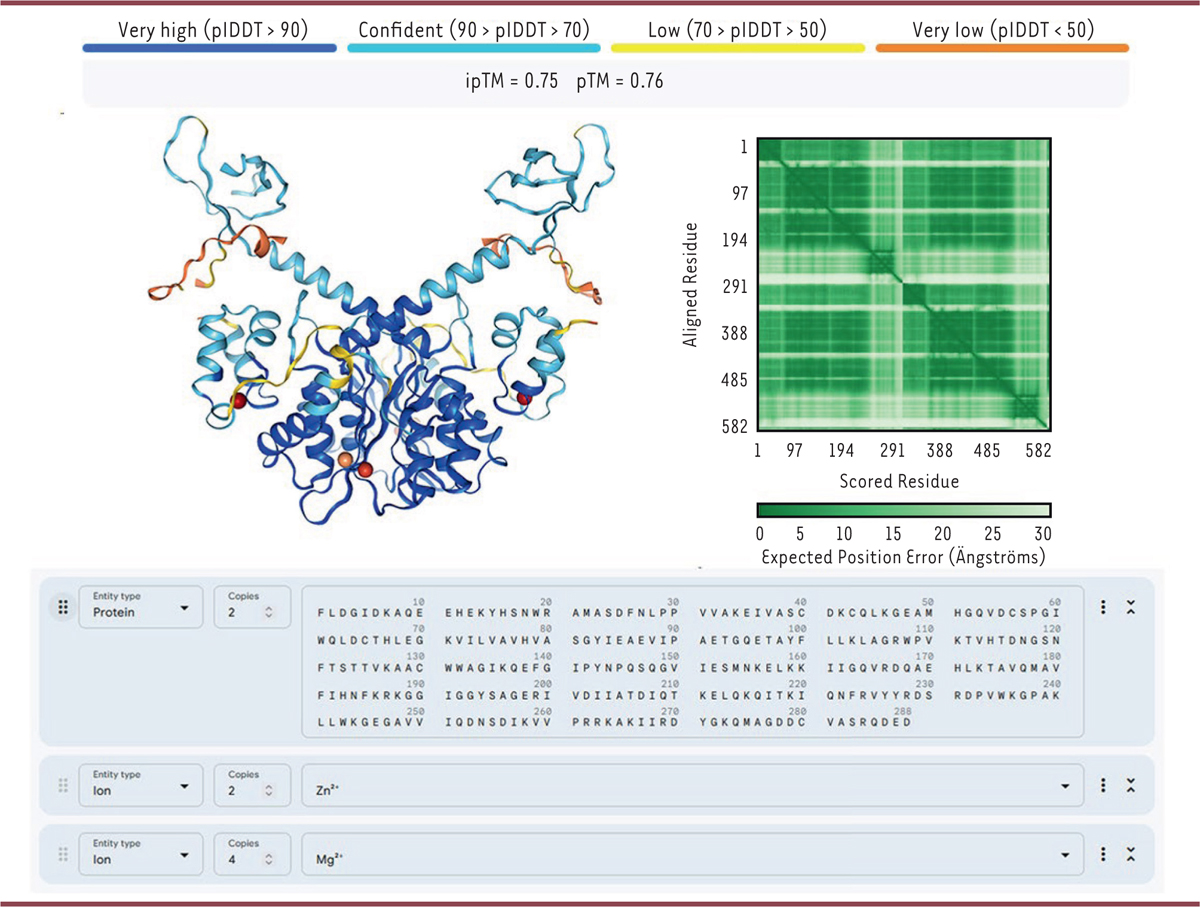
Structure homodimérique de l’intégrase (IN) du virus VIH-1 (en haut à gauche) prédite par AlphaFold-3 à partir de sa séquence et d’ions liés (panneau bleu en bas). La protéine est représentée en mode ruban et est colorée en fonction de son score de confiance local plDDT. Cette protéine modulaire de 288 résidus est constituée de trois domaines : un domaine amino-terminal (NTD, 1-47) un domaine central catalytique (CCD, 59-202) et un domaine carboxy-terminal (CTD, 220-270) [39]. Le NTD lie un ion Zn2+ et le CCD deux ions Mg2+. Les structures des fragments NTD-CCD et CCD-CTD ont été résolues expérimentalement sous forme d’homodimère pour l’IN seule. La structure prédite de l’IN homodimérique entière montre de bonnes statistiques globales avec un pTM (predicted template modeling score) de 0.76 (>0.5 attendu) et un ipTM (interface pTM-score) de 0.75 (>0.6 attendu). L’analyse du PAE (predicted aligned error) (en haut à droite) montre une bonne rigidité au sein de chaque domaine (zones vert sombre) ainsi qu’entre les domaines CCD des deux monomères. La position du CTD par rapport au CCD et au NTD est légèrement flexible (zone vert clair) au sein de chaque monomère. La boucle reliant le NTD au CCD et la queue carboxy-terminale (271-288) sont plus flexibles (zones quasi-blanche). Ceci est confirmé par les couleurs jaune et orange de ces deux segments sur la représentation de la protéine en mode ruban (score de confiance plDTT localement bas à très bas).
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.