")
")
| Issue |
Med Sci (Paris)
Volume 40, Number 8-9, Août-Septembre 2024
Chroniques génomiques
|
|
|---|---|---|
| Page(s) | 677 - 679 | |
| Section | Forum | |
| DOI | https://doi.org/10.1051/medsci/2024093 | |
| Published online | 20 September 2024 | |
Thérapie génique in vivo : une approche modulaire
A modular approach to in vivo gene therapy
Biologiste, généticien et immunologiste, Président d’Aprogène (Association pour la promotion de la Génomique), 13007 Marseille, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Abstract
In vivo inactivation of a deleterious gene has been achieved in a small trial, with excellent clinical results. Interestingly, the delivery and editing system is the same as in previous work on a different disease, and the new therapy required simply changing the guide RNA used to target the Cas9 nuclease. This modular approach could be extended to a number of other genetic diseases.
© 2024 médecine/sciences – Inserm
 Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l’utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l’utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.

Une affection génétique rare mais invalidante
L’édition du génome par le système CRISPR-Cas9 est en passe de révolutionner les approches de thérapie génique [1] (→), et l’on vient d’enregistrer un nouveau succès pour sa version in vivo, sans doute la plus prometteuse. J’avais déjà rapporté des résultats positifs (mais un peu préliminaires) pour cette approche appliquée à l’amylose à transthyrétine héréditaire [2] (→).
(→) Voir l’Éditorial de H. Chneiweiss, m/s n° 8-9, août-septembre 2023, page 589
(→) Voir la Chronique génomique de B. Jordan, m/s n° 10, octobre 2021, page 933
Cette fois, il s’agit de traiter l’angio-œdème (ou œdème de Quinke) héréditaire, maladie rare (1 cas pour 67 000 personnes) mais très handicapante, qui se transmet sur le mode autosomique dominant [3]. Les patients subissent des attaques qui peuvent être fréquentes (plus d’une dizaine par mois), durent de quelques heures à quelques jours, et consistent en un œdème souvent défigurant (Figure 1) [4], et qui peut être dangereux s’il touche les tissus du larynx.
Le mécanisme le plus fréquent est une mutation du gène C1NH (C1 inhibitor gene) provoquant un déficit en inhibiteur de C1 (C1inh)1, produit dans le foie. Cela augmente la production de kallicréine (une protéase à sérine intervenant dans les mécanismes de l’inflammation, gène KLKB1) ce qui augmente la perméabilité vasculaire et provoque l’œdème via une cascade enzymatique. On peut traiter (ou du moins atténuer) l’angio-œdème en supplémentant le malade en inhibiteur de C1 [5] ou en inhibant la kallikréine : cette protéine constitue une cible thérapeutique cliniquement validée. Un anticorps monoclonal dirigé contre la kallikréine, appelé Lanadelumab et administré en injection sous-cutanée2 a démontré une certaine efficacité [6] (réduction de 75 % de la fréquence des attaques), mais il doit être administré toutes les deux semaines et à vie. Une inhibition du gène KLKB1 a aussi été tentée grâce à un oligonucléotide antisens [7], avec des résultats plus convaincants (fréquence des attaques divisée par dix), mais là aussi, il s’agit d’un traitement à poursuivre indéfiniment3. Une thérapie génique inactivant le gène KLKB1 serait largement préférable, c’est ce que présente l’article que je vais analyser.
Une approche déjà explorée avec un certain succès
J’avais déjà rapporté les résultats d’une tentative de thérapie génique in vivo menée par l’entreprise Intellia en collaboration avec des équipes universitaires [2, 8] dans le but de traiter une amylose héréditaire. Il s’agissait dans ce cas d’inactiver le gène TTR de la transthyrétrine, protéine exprimée dans le foie et dont la surproduction cause cette affection. Les auteurs avaient construit un système de type CRISPR-Cas9 dans lequel des nanoparticules lipidiques contenant l’ARN messager de la protéine Cas9 et un ARN guide dirigé vers le gène TTR étaient injectées au malade par voie intraveineuse. Ces nanoparticules étaient dirigées vers le foie par leurs lipides de surface, et provoquaient la synthèse d’un complexe Cas9/ARN guide qui allait alors pratiquer une coupure double brin à l’emplacement choisi de l’ADN du gène TTR. Cette coupure, suivie d’une réparation imparfaite (comme c’est la règle en cas de coupure double brin), devait aboutir à l’inactivation du gène TTR. Les auteurs avaient effectivement observé lors d’un essai de phase I une réduction drastique de la concentration sérique de la protéine TTR après traitement avec cet agent baptisé NTLA-2001, mais ne présentaient pas de données cliniques sur l’amélioration des symptôme [8]. Un essai de phase III est actuellement en cours, mais il n’y a pas encore de publication. Ce premier essai, encore incomplet, était néanmoins très encourageant.
De NTLA-2001 à NTLA-2002 : une tentative réussie et un cas d’école
Le travail rapporté ici [9] implique pour l’essentiel les mêmes équipes, majoritairement l’industriel Intellia (17 auteurs sur les 21 de l’article) associé à des équipes des États-Unis, du Royaume-Uni et de Nouvelle Zélande. L’objectif était d’inactiver le gène de la kallikréine (KLKB1) afin de faire baisser d’au moins 60 % le niveau de cette protéine. Cet objectif ne comporte pas de risque avéré pour les malades puisqu’il a été montré qu’une déficience totale en kallikréine ne provoque pas de trouble particulier à part un retard modéré de coagulation sanguine[10]. Les auteurs ont utilisé le même système que précédemment : il a suffi de changer l’ARN guide afin qu’il cible une séquence du gène KLKB1 au lieu de TTR, la kallikréine étant elle aussi exprimée dans le foie. Bien entendu des vérifications préliminaires ont été menées afin de s’assurer de la spécificité du système (pas de coupure hors cible [offtarget]). L’essai de ce produit, baptisé NTLA-2002, a porté sur dix patients (seulement – l’affection est très rare) et constitue une phase I-II sans placebo, loin de l’idéal de l’essai randomisé en double aveugle avec placebo4, mais ses résultats n’en sont pas moins solides et importants. On observe en effet, dans les semaines qui suivent le traitement (une injection intraveineuse unique de NTLA-002), une baisse spectaculaire du niveau sérique de la kallikréine, supérieure à 60 % dans tous les cas et atteignant même 90 % pour la dose la plus élevée de NTLA-2002 (Figure 2).
 |
Figure 2. Réduction du niveau sérique de kallikréine au cours des semaines suivant un traitement unique par NTLA-2002 à différentes doses (indiquées en haut). Les barres d’erreurs sont importantes en raison du faible effectif de l’essai (extrait modifié de la figure 1 de [9]). |
Contrairement à l’article précédent portant sur le gène TTR et l’agent NTLA-2001, cet article comporte également des résultats cliniques, qui sont eux aussi spectaculaires. Les patients recrutés pour l’essai présentaient un historique d’attaques d’œdème fréquentes (de 3 à 54 au cours du trimestre précédent l’essai), et étaient pour la plupart sous traitement par inhibiteur de la kallikréine. Dans les semaines suivant le traitement, les attaques ont diminué, puis quasiment disparu, et tous les malades ont arrêté leur traitement au bout de trois à quatre mois sans que cela ne provoque de rechute. La Figure 3 montre deux exemples : un patient sévèrement atteint, dont le nombre élevé d’attaques se réduit puis disparaît au bout de quelques semaines, et un patient plus légèrement atteint qui semble guéri dès l’injection.
 |
Figure 3. Évolution de l’affection après thérapie génique chez deux patients (75-01 et 75-02) traités par une dose de 75 mg de NTLA-2002. Le nombre moyen d’attaques par mois avant traitement est indiqué, il est très élevé pour le premier patient (75-01 : 16,8), il diminue drastiquement dès le traitement (indiqué par la barre verticale noire) de même que la sévérité des attaques (traits verticaux de couleur), codée de vert (faible) à rouge (très intense). Bien que toujours présentes, les attaques sont de moindre intensité (traits verts comparés aux traits rouges avant traitement). Pour le deuxième patient, les attaques disparaissent complétement dès le traitement. Les lettres « X » repèrent la semaine 16 après traitement (extrait partiel et modifié de la figure 2 de [9]). |
Les effets secondaires du traitement se sont avérés très modérés (réactions à l’injection, fatigue), et l’efficacité globale pour la prévention des crises ressort à 95 % en combinant tous les patients, ce qui est excellent. La réduction du niveau de kallikréine dépend de la dose de NTLA-2002, mais l’effet clinique ne semble pas être lié à la dose utilisée : la réduction du nombre d’attaques est de 95 % pour les malades traités par une dose de 25 mg, de 98 % pour 50 mg, et de 93 % pour 75 mg. Il serait peut-être possible d’utiliser des doses plus faibles. En tous cas, ces résultats sont spectaculaires et constituent un cas d’école pour la thérapie génique in vivo, même si, bien sûr, ils doivent être confirmés par des études plus larges et un suivi de longue durée.
Une approche modulaire
Ce succès s’ajoute aux autres réussites récentes de thérapie génique in vivo, ne supposant pas l’obtention de cellules à modifier ex vivo avant de les réintroduire dans l’organisme du patient [11]. Ce qui semblait un rêve inaccessible il y a quelques années devient réalité, grâce à l’utilisation intelligente du système CRISPR-Cas 9 et à de grands progrès dans la mise au point de vecteurs pour transporter les réactifs. On ne peut, de plus, qu’être frappé par la simplicité du passage d’une thérapie de l’amylose héréditaire à celle de l’angio-œdème : il a suffi de modifier la séquence de l’ARN guide contenu dans les nanoparticules lipidiques pour obtenir un nouveau traitement, dont l’efficacité s’est avérée excellente. L’approche est a priori applicable à toute thérapie dont l’objectif est d’inactiver un gène exprimé dans le foie : il ne faudra qu’un changement de l’ARN guide pour obtenir le nouveau traitement NTLA-200X ! Notons cependant que, contrairement à ce qu’indique le titre de l’article [9]5, il ne s’agit pas à proprement parler d’édition du génome, c’est-à-dire d’une modification précise en un point d’un gène, mais de l’inactivation de ce dernier par coupure suivie de réparation imparfaite : il est plus facile techniquement d’inactiver un gène que de le modifier de manière précise. Mais c’est déjà possible, comme le montre un tout récent article [12] qui rapporte la correction d’une mutation ponctuelle dans le gène CEP290 (centrosomal protein, 290-Kd), cause d’une perte progressive de vision : l’essai de (véritable) édition in vivo sur un tout petit nombre de malades a montré une amélioration significative de leur vision. Les succès se multiplient. Reste à savoir si ce traitement, une fois validé, sera accessible pour les malades. S’agissant d’une maladie très rare, dont le traitement actuel est imparfait et très coûteux6, on peut s’attendre à un tarif de l’ordre du million d’euros, « justifié » par le traitement définitif d’une affection dont la médication actuelle coûte plus de cent mille euros par année. Un tarif aussi élevé permet à l’entreprise (Intellia dans le cas présent) d’amortir ses coûts de recherche et de réaliser un profit ; la très grande rareté de l’affection limitera la charge financière de cette thérapie pour les systèmes de santé. Mais l’avènement de thérapies géniques (in vivo ou ex vivo) efficaces pour des affections relativement fréquentes, comme la mucoviscidose, la myopathie de Duchenne ou l’anémie falciforme, va poser de manière aiguë la question du coût de ces traitements, comme c’est déjà le cas pour la thérapie de l’hépatite C [13]7.
Liens d’intérêt
L’auteur déclare n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Inhibiteur de la C1 estérase qui participe à la régulation de la voie classique du complément.
Récemment autorisé en France sous le nom de Takhzyro, au prix de près de 12 000 € pour une dose à renouveler toutes les deux semaines.
Baptisé donidalorsen, ce produit est en cours d’étude par la FDA (Food and Drug Administration) en vue de sa mise sur le marché.
CRISPR-Cas9 In Vivo Gene Editing of KLKB1 for Hereditary Angioedema.
12 000,00 euros l’injection bimensuelle de Takhzyro, anticorps monoclonal antikallikréine.
Le coût du traitement de l’hépatite C en France est d’environ 30 000 € (pris en charge à 100 %).
Références
- Chneiweiss H. 2023 : premiers succès et nouveaux enjeux de l’édition du génome en thérapeutique humaine. Med Sci (Paris) 2023 ; 39 : 589–90. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Jordan B. Édition de gènes in vivo et thérapie génique Med Sci (Paris) 2021 ; 37 : 933–5. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Bernstein JA. Severity of hereditary angioedema, prevalence, and diagnostic considerations. Am J Manag Care 2018 ; 24 : S292–8. [PubMed] [Google Scholar]
- Ebo DG, Bridts CH. Images in clinical medicine. Disfiguring angioedema. N Engl J Med 2012 ; 367 : 1539. [CrossRef] [PubMed] [Google Scholar]
- Zuraw BL, Busse PJ, White M, et al. Nanofiltered C1 inhibitor concentrate for treatment of hereditary angioedema. N Engl J Med 2010 ; 363 : 513–22. [CrossRef] [PubMed] [Google Scholar]
- Banerji A, Riedl MA, Bernstein JA, et al. Effect of lanadelumab compared with placebo on prevention of hereditary angioedema attacks: a randomized clinical trial. JAMA 2018 ; 20 : 2108–21. [CrossRef] [PubMed] [Google Scholar]
- Fijen LM, Riedl MA, Bordone L, et al. Inhibition of Prekallikrein for Hereditary Angioedema. N Engl J Med 2022 ; 386 : 1026–33. [CrossRef] [PubMed] [Google Scholar]
- Gillmore JD, Gane E, Taubel J, et al. CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. N Engl J Med 2021 ; 385 : 493–502. [CrossRef] [PubMed] [Google Scholar]
- Longhurst HJ, Lindsay K, Petersen RS, et al. CRISPR-Cas9 In Vivo Gene Editing of KLKB1 for Hereditary Angioedema. N Engl J Med 2024 ; 390 : 432–41. [CrossRef] [PubMed] [Google Scholar]
- Barco S, Sollfrank S, Trinchero A, et al. Severe plasma prekallikrein deficiency: clinical characteristics, novel KLKB1 mutations, and estimated prevalence. J Thromb Haemost 2020 ; 18 : 1598–617. [CrossRef] [PubMed] [Google Scholar]
- Henderson ML, Zieba JK, Li X, et al. Gene Therapy for Genetic Syndromes: Understanding the Current State to Guide Future Care. BioTech (Basel) 2024 ; 13 : 1. [CrossRef] [Google Scholar]
- Pierce EA, Aleman TS, Jayasundera KT, et al. Gene Editing for CEP290-Associated Retinal Degeneration. N Engl J Med 2024. doi: 10.1056/NEJMoa2309915. [PubMed] [Google Scholar]
- Henry B. Drug Pricing & Challenges To Hepatitis C Treatment Access. J Health Biomed Law 2018 : 14 : 265–83. [PubMed] [Google Scholar]
Liste des figures
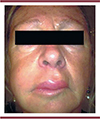 |
Figure 1. Patient lors d’une crise d’angio-œdème [4]. |
| Dans le texte | |
|
Figure 2. Réduction du niveau sérique de kallikréine au cours des semaines suivant un traitement unique par NTLA-2002 à différentes doses (indiquées en haut). Les barres d’erreurs sont importantes en raison du faible effectif de l’essai (extrait modifié de la figure 1 de [9]). |
| Dans le texte | |
|
Figure 3. Évolution de l’affection après thérapie génique chez deux patients (75-01 et 75-02) traités par une dose de 75 mg de NTLA-2002. Le nombre moyen d’attaques par mois avant traitement est indiqué, il est très élevé pour le premier patient (75-01 : 16,8), il diminue drastiquement dès le traitement (indiqué par la barre verticale noire) de même que la sévérité des attaques (traits verticaux de couleur), codée de vert (faible) à rouge (très intense). Bien que toujours présentes, les attaques sont de moindre intensité (traits verts comparés aux traits rouges avant traitement). Pour le deuxième patient, les attaques disparaissent complétement dès le traitement. Les lettres « X » repèrent la semaine 16 après traitement (extrait partiel et modifié de la figure 2 de [9]). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.