")
")
| Issue |
Med Sci (Paris)
Volume 40, Number 5, Mai 2024
|
|
|---|---|---|
| Page(s) | 402 - 404 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/2024042 | |
| Published online | 31 May 2024 | |
L’administration locale d’un vecteur AAV-TMPRSS3 à un âge avancé corrige le déficit auditif progressif dans un modèle murin de la surdité DFNB8
Local administration of an AAV-TMPRSS3 vector in aged Tmprss3 mutant mice with progressive deafness restores hearing
Mécanismes moléculaires dans les démences neurodégénératives, Université de Montpellier, École pratique des hautes études, Inserm U1198, Montpellier, France
* This email address is being protected from spambots. You need JavaScript enabled to view it.
Un à deux enfants sur 1 000 naissent avec une surdité profonde, et un enfant sur 700 développera une surdité avant l’âge adulte [1]. Près de 80 % des surdités congénitales sont d’origine génétique. La plupart de ces surdités résultent de la perte irréversible de différents types de cellules de la cochlée : les cellules ciliées internes, qui sont les véritables cellules sensorielles transformant l’onde sonore en message nerveux ; les cellules ciliées externes, qui ont un rôle mécanique amplificateur ; les neurones auditifs primaires, qui véhiculent le message nerveux jusqu’au noyau cochléaire du tronc cérébral, premier relai des voies sensorielles de l’audition ; et les cellules de la strie vasculaire, une structure épithéliale cochléaire produisant l’endolymphe, le fluide dans lequel baigne la partie apicale des cellules de l’épithélium sensoriel (Figure 1). L’analyse des formes de surdité héréditaire offre une opportunité unique d’identifier des gènes essentiels pour l’audition. À ce jour, 105 locus de surdité récessive (DFNB) et 79 locus de surdité dominante (DFNA) ont été décrits dans le génome humain (sans compter ceux, encore plus nombreux, des surdités syndromiques1), et la plupart des gènes responsables ont été identifiés. Les protéines correspondantes sont classifiées selon leur fonction 1) dans la structure ou le fonctionnement de la touffe ciliaire coiffant les cellules ciliées, 2) dans l’homéostasie ionique de la cochlée, 3) dans la composition de la matrice extracellulaire, ou 4) dans le contrôle de l’expression génique.
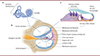 |
Figure 1. La cochlée, organe de l’audition. A. L’oreille interne des mammifères comprend la cochlée, organe sensoriel de l’audition, et cinq organes sensoriels vestibulaires pour l’équilibration. B. Une coupe transversale de la cochlée montre ses trois canaux membraneux juxtaposés: les scala vestibuli et tympani, remplies de la périlymphe, et la scala media ou canal cochléaire, remplie de l’endolymphe. Le canal cochléaire est séparé de la scala vestibuli par la membrane de Reissner et de la scala tympani par la membrane basilaire. L’épithélium sensoriel auditif, ou organe de Corti, fait saillie dans le canal cochléaire. Il est surmonté d’un gel acellulaire, la membrane tectoriale. Il est composé des cellules ciliées et de leurs cellules de soutien, dont la partie apicale (au-dessus des jonctions intercellulaires serrées) baigne dans l’endolymphe, et le reste dans la périlymphe. La composition ionique de l’endolymphe, très différente de celle des autres fluides extracellulaires de l’organisme (incluant la périlymphe), est proche de celle du milieu intracellulaire: riche en ions K+ (environ 150mM) et pauvre en ions Na+ (1mM). Par ailleurs, il existe une différence transépithéliale de potentiel électrique (environ +100mV) entre les compartiments endolymphatique et périlymphatique de la cochlée, qui contribue majoritairement à la force motrice du courant ionique de la transduction mécano-électrique. Les cellules ciliées sont innervées par les neurones du ganglion spiralé. C. Un agrandissement de l’organe de Corti montre les deux types de cellules ciliées: les cellules ciliées internes, qui sont les véritables cellules sensorielles auditives, et les cellules ciliées externes, qui ont un rôle mécanique d’amplification locale de l’oscillation de l’organe de Corti au passage de l’onde sonore grâce à leur propriété d’électromotilité. |
TMPRSS3 est une protéase à sérine transmembranaire de type II du réticulum endoplasmique, qui comprend un domaine transmembranaire, un domaine LDLRA (low density lipoprotein receptor A), un domaine SRCR (scavenger receptor cysteine-rich) et un domaine SP (serine protease). Le gène TMPRSS3 est exprimé dans différents types cellulaires de la cochlée (cellules de la strie vasculaire, cellules ciliées, neurones auditifs primaires), et la protéine active in vitro le canal sodique épithélial ENaC [2]. Des variants de TMPRSS3 sont responsables des surdités récessives DFNB8 et DFNB10 [3]. DFNB8 est une surdité postlinguale2 progressive, alors que DFNB10 est une surdité sévère d’apparition précoce, prélinguale. Plusieurs dizaines de mutations différentes de TMPRSS3 ont été décrites dans plus de vingt groupes ethniques [4]. À ce jour, la seule option de réhabilitation auditive pour ces surdités est la pose chirurgicale d’un implant cochléaire3. Toutefois, les résultats varient en fonction notamment de l’âge d’implantation, mais également de l’atteinte initiale [5]. De plus, cette technique ne résout pas les difficultés de perception des sons dans un environnement bruyant.
L’utilisation de vecteurs viraux à des fins de thérapie génique a connu un essor spectaculaire ces deux dernières décennies. Dans la perspective d’une application de cette thérapie à certaines formes génétiques de surdité, les virus adéno-associés (AAV) ont rapidement été privilégiés du fait de leur capacité à transduire efficacement et durablement les cellules cibles dans l’oreille interne, assurant une expression du transgène tout en induisant une réaction immunitaire limitée. Les cellules ciblées par les AAV dépendent de leur sérotype, c’est pourquoi le choix de celui-ci est primordial pour l’élaboration de la stratégie thérapeutique. Enfin, la voie d’administration doit permettre une bonne distribution des particules virales tout en préservant l’intégrité de l’oreille interne [6, 7] (→).
(→) Voir la Synthèse de A. Meyer et al., m/s n° 10, octobre 2013, page 883
La majorité des études publiées concernant la thérapie génique des surdités ont utilisé des modèles murins à des âges post-natals précoces (allant du stade nouveau-né à juvénile), auxquels la cochlée n’était pas encore mature. Cependant, chez l’homme, le développement cochléaire se déroule avant la naissance. Quant à la surdité DFNB8, elle est d’apparition tardive : il était donc important d’apporter une preuve de concept quant à l’efficacité de la transduction lors d’une injection de particules virales dans un modèle animal à un âge où la cochlée est mature. À cet effet, une équipe de chercheurs a généré une souris portant la mutation faux-sens c.916G>A (p.Ala306Thr) responsable de la surdité tardive progressive DFNB8 chez l’homme [8]. Contrairement au modèle murin Tmprss3 Y260X/Y260X de la surdité humaine DFNB10, qui présente une surdité congénitale profonde [9], la souris mutante Tmprss3 A306T/A306T a une audition comparable à celle de souris témoins jusqu’à l’âge de 10,5 mois. Cependant, à partir de 16,5 mois, on constate une augmentation des seuils de perception à l’enregistrement des potentiels évoqués auditifs du tronc cérébral (accompagnée d’une diminution de l’amplitude de l’onde 1 reflétant l’activité électrique du nerf auditif) et des seuils de recueil des otoémissions acoustiques. Cette surdité progressive est associée à des pertes de cellules ciliées internes et externes, ainsi qu’à une dégénérescence des neurones auditifs primaires [8].
Le transfert, à des souris témoins de génotype « sauvage » âgées de 2 mois, du gène Tmprss3 murin au moyen de vecteurs AAV2, par la fenêtre ronde de la cochlée après perforation du canal cochléaire, s’est révélée fortement ototoxique, entraînant une surdité sévère et une perte des cellules ciliées externes. Étonnamment, l’injection du gène TMPRSS3 humain, malgré une homologie de séquence nucléotidique de 89 % avec le gène murin, n’a, quant à elle, induit aucune perte auditive chez les souris témoins, validant ainsi une utilisation des particules virales hTMPRSS3-AAV2 dans un but thérapeutique. Chez les souris mutantes Tmprss3 A306T/A306T, les auteurs ont alors fait le choix de réaliser une injection unique, unilatérale (l’autre oreille étant utilisée comme témoin interne), des particules hTMPRSS3-AAV2 à l’âge de 18,5 mois, soit deux mois après la première atteinte auditive constatée chez ces souris. Cette injection a permis une transduction efficace des cellules ciliées et des neurones du ganglion spiralé, ainsi que la préservation de ces cellules chez les souris mutantes. Sur le plan fonctionnel, la présence de la protéine TMPRSS3 humaine dans ces cellules a restauré les seuils des potentiels évoqués auditifs (avec augmentation de l’amplitude de l’onde 1) et des otoémissions acoustiques pour l’oreille injectée. L’effet thérapeutique de la transduction des cellules par le vecteur viral a été évalué régulièrement jusqu’à quatre mois après l’injection de ce vecteur (i.e., jusqu’à l’âge de 22,5 mois), date à laquelle l’effet bénéfique de l’expression de hTMPRSS3 sur l’audition des souris mutantes était toujours présent [8] (Figure 2). Toutefois, il convient de souligner la limite de ce modèle murin sur fonds génétique CBA/CaJ, car ces souris développent spontanément une surdité liée au vieillissement, qui empêche de prolonger l’analyse longitudinale des effets de cette thérapie génique tardive.
 |
Figure 2. Schéma du protocole de thérapie génique de la surdité murine par mutation faux-sens de Tmprss3. En haut: les souris mutantes Tmprss3A306T/A306T (modèle murin de la surdité humaine DFNB8) ont une perte auditive progressive à partir de l’âge de 16,5 mois, associée à une dégénérescence des cellules ciliées de l’épithélium sensoriel de la cochlée. En bas: l’administration locale d’un vecteur viral AAV2 codant la protéine TMPRRS3 humaine à ces souris à l’âge de 18,5 mois a corrigé cette perte auditive et préservé les cellules ciliées plus de quatre mois après l’injection thérapeutique. |
Cette étude a montré l’efficacité d’une thérapie génique tardive sur une cochlée mature, et ouvre ainsi de nouvelles perspectives d’un traitement curatif pour les personnes atteintes de la surdité DFNB8 due à des mutations faux-sens de TMPRSS3. Les résultats prometteurs obtenus avec un modèle murin de cette forme de surdité devraient en effet encourager la réalisation d’un essai thérapeutique chez l’homme. Un tel essai s’inscrira dans le cadre de programmes internationaux, dont l’un a récemment fait état du succès thérapeutique de l’injection locale du gène OTOF à des personnes atteintes de la surdité DFNB9, due à un défaut de l’exocytose des vésicules synaptiques des cellules ciliées internes4. Dans le cas de la surdité progressive DFNB8, si le diagnostic est posé suffisamment tôt, la thérapie génique pourrait préserver les cellules ciliées et les neurones du ganglion spiralé de la dégénérescence induite par l’absence de protéine TMPRSS3 fonctionnelle. Si la surdité est plus avancée, la thérapie génique pourra être envisagée en combinaison d’un implant cochléaire, afin de protéger les neurones du ganglion spiralé encore fonctionnels.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Les surdités syndromiques sont celles qui s’accompagnent d’anomalies du développement d’autres organes, en dehors du système auditif. Les surdités DFNA et DFNB désignent des surdités isolées (non syndromiques).
Ce terme désigne une surdité qui apparaît après l’âge d’acquisition du langage parlé.
Dispositif électronique implanté chez des enfants (ou des adultes) atteints d’une surdité sévère ou profonde, qui permet de rétablir une audition en court-circuitant le traitement du son par la cochlée défectueuse : des électrodes posées chirurgicalement permettent de stimuler directement les terminaisons nerveuses des fibres du nerf auditif.
47e congrès annuel de l’Association for Research in Otolaryngology, 2024, Anaheim, États-Unis.
Références
- Petit C. From deafness genes to hearing mechanisms: harmony and counterpoint. Trends Mol Med 2006 ; 12 : 57–64. [CrossRef] [PubMed] [Google Scholar]
- Guipponi M, Vuagniaux G, Wattenhofer M, et al. The transmembrane serine protease (TMPRSS3) mutated in deafness DFNB8/10 activates the epithelial sodium channel (ENaC) in vitro. Hum Mol Genet 2002 ; 11 : 2829–2836. [CrossRef] [PubMed] [Google Scholar]
- Scott HS, Kudoh J, Wattenhofer M, et al. Insertion of β-satellite repeats identifies a transmembrane protease causing both congenital and childhood onset autosomal recessive deafness. Nat Genet 2001 ; 27 : 59–63. [CrossRef] [PubMed] [Google Scholar]
- Moon IS, Grant AR, Sagi V, et al. TMPRSS3 gene variants with implications for auditory treatment and counseling. Front Genet 2021; 12 : 780874. [CrossRef] [PubMed] [Google Scholar]
- Fehrmann MLA, Huinck WJ, Thijssen MEG, et al. Stable long-term outcomes after cochlear implantation in subjects with TMPRSS3-associated hearing loss: a retrospective multicentre study. J Otolaryngol Head Neck Surg 2023; 52 : 82. [CrossRef] [PubMed] [Google Scholar]
- Hahn R, Avraham KB. Gene therapy for inherited hearing loss: updates and remaining challenges. Audiol Res 2023; 13 : 952–66. [CrossRef] [PubMed] [Google Scholar]
- Meyer A, Petit C, Safieddine S. Thérapie génique des surdités humaines : défis et promesses. Med Sci (Paris) 2013 ; 29 : 883–889. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Du W, Ergin V, Loeb C, et al. Rescue of auditory function by a single administration of AAV-TMPRSS3 gene therapy in aged mice of human recessive deafness DFNB8. Mol Ther 2023 ; 31 : 2796–810. [CrossRef] [PubMed] [Google Scholar]
- Fasquelle L, Scott HS, Lenoir M, et al. Tmprss3, a transmembrane serine protease deficient in human DFNB8/10 deafness, is critical for cochlear hair cell survival at the onset of hearing. J Biol Chem 2011 ; 286 : 17383–97. [CrossRef] [PubMed] [Google Scholar]
© 2024 médecine/sciences – Inserm
 Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l'utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l'utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Liste des figures
|
Figure 1. La cochlée, organe de l’audition. A. L’oreille interne des mammifères comprend la cochlée, organe sensoriel de l’audition, et cinq organes sensoriels vestibulaires pour l’équilibration. B. Une coupe transversale de la cochlée montre ses trois canaux membraneux juxtaposés: les scala vestibuli et tympani, remplies de la périlymphe, et la scala media ou canal cochléaire, remplie de l’endolymphe. Le canal cochléaire est séparé de la scala vestibuli par la membrane de Reissner et de la scala tympani par la membrane basilaire. L’épithélium sensoriel auditif, ou organe de Corti, fait saillie dans le canal cochléaire. Il est surmonté d’un gel acellulaire, la membrane tectoriale. Il est composé des cellules ciliées et de leurs cellules de soutien, dont la partie apicale (au-dessus des jonctions intercellulaires serrées) baigne dans l’endolymphe, et le reste dans la périlymphe. La composition ionique de l’endolymphe, très différente de celle des autres fluides extracellulaires de l’organisme (incluant la périlymphe), est proche de celle du milieu intracellulaire: riche en ions K+ (environ 150mM) et pauvre en ions Na+ (1mM). Par ailleurs, il existe une différence transépithéliale de potentiel électrique (environ +100mV) entre les compartiments endolymphatique et périlymphatique de la cochlée, qui contribue majoritairement à la force motrice du courant ionique de la transduction mécano-électrique. Les cellules ciliées sont innervées par les neurones du ganglion spiralé. C. Un agrandissement de l’organe de Corti montre les deux types de cellules ciliées: les cellules ciliées internes, qui sont les véritables cellules sensorielles auditives, et les cellules ciliées externes, qui ont un rôle mécanique d’amplification locale de l’oscillation de l’organe de Corti au passage de l’onde sonore grâce à leur propriété d’électromotilité. |
| Dans le texte | |
|
Figure 2. Schéma du protocole de thérapie génique de la surdité murine par mutation faux-sens de Tmprss3. En haut: les souris mutantes Tmprss3A306T/A306T (modèle murin de la surdité humaine DFNB8) ont une perte auditive progressive à partir de l’âge de 16,5 mois, associée à une dégénérescence des cellules ciliées de l’épithélium sensoriel de la cochlée. En bas: l’administration locale d’un vecteur viral AAV2 codant la protéine TMPRRS3 humaine à ces souris à l’âge de 18,5 mois a corrigé cette perte auditive et préservé les cellules ciliées plus de quatre mois après l’injection thérapeutique. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.