")
")
Figure 3.
Télécharger l'image originale
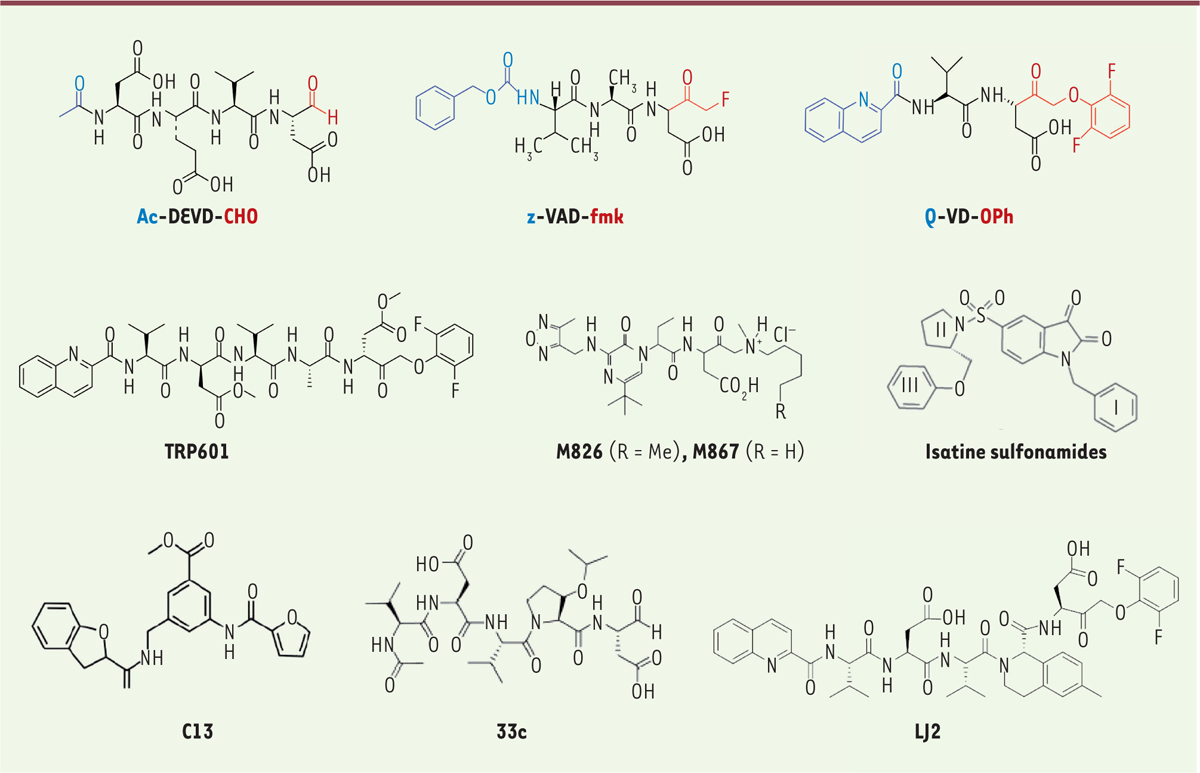
Structure de quelques inhibiteurs de caspases. Ac-DEVD-CHO, est un tétrapeptide aldéhyde qui inhibe préférentiellement et de manière réversible les Caspase-3 (Ki = 0,23nM) et Caspase-8 (Ki = 0,92nM). Sa pénétration cellulaire est très mauvaise, comme sa biodisponibilité, ce qui en fait une molécule inenvisageable pour une utilisation comme médicament. z-VAD-fmk est un inhibiteur irréversible qui (sous sa forme déméthylée en P1) présente un spectre large contre des caspases (à l’exception de la Caspase-2) et l’inhibition croisée de plusieurs cathepsines. Le groupe partant CH2F (FMK) génère in vivo un fluoroacétate qui est hautement toxique. Q-VD-OPh est un dérivé de dipeptide, inhibiteur irréversible pan-caspase de troisième génération [38]. Son extrémité N-ter lui confère une bonne biodisponibilité et son groupe partant est puissant. Il n’inhibe pas d’enzymes hors de la famille des caspases. Un composé proche du Q-VD-OPh, l’emricasan, compatible avec une administration per os, a été développé chez l’homme jusqu’en phase III (voir Figure 4). TRP601 est un dérivé pentapeptidique du Q-VD-OPh, incluant une séquence VD(Ome)VAD(Ome). Son métabolite actif le Δ2Me-TRP601 (déméthylation des 2 aspartates) est un inhibiteur puissant préférentiel des caspases du groupe II). Il a été développé par la biotech française Theraptosis contre les lésions cérébrales néonatales puis cédé à Chiesi Pharmaceuticals à la fin de son développement préclinique [64]. M826, M827 sont des inhibiteurs non peptidiques, sélectifs et réversibles de Caspase-3, présentant une très forte activité anti-apoptotique in vitro et in vivo. Ils proviennent d’une série dans laquelle le squelette peptidique P4-P3-P2 a été remplacé par un module amino-pyrazinone. Les composés M867 et M826 présentent une affinité sub-nanomolaire vis-à-vis de la Caspase-3 (Ki ~ 0,7 nM) et une bonne sélectivité en regard des autres caspases. Les deux composés sont très efficaces dans des modèles cellulaires d’apoptose (20 nM < IC50 < 100 nM). Ces composés et leur dérivé M791 (non montré) sont les inhibiteurs les plus puissants et sélectifs de Caspase-3 découverts à ce jour. Les isatines sulfonamides sont des séries chimiques qui ont été très étudiées pour l’inhibition des caspases. Sur la base de la structure présentée ici, de nombreux variants (modifications des domaines I, II, et III) ont été synthétisés et évalués. Certains sont de très bons inhibiteurs des Capases-3 et -7. L’une des difficultés de ses structures est de combiner efficacité et stabilité chimique. C13 est un inhibiteur allostérique de Caspase-6 (Ki~2 µM), obtenu après criblage virtuel de 57 700 composés pour leur interaction avec la poche allostérique putative de la Caspase-6 [54]. 33c est un inhibiteur réversible préférentiel de Caspase-2 (Ki = 122nM) obtenu à partir du pentapeptide VDVAD par le remplacement du résidu en position P2 par un synthon non naturel qui gêne l’interaction avec le site S2 de la Caspase-3 mais reste compatible avec celui de la Caspase-2 [46]. Son proche dérivé 33 h présente une affinité améliorée pour la Caspase-2 (Ki = 10,9nM) et est 600 fois moins affin pour la Caspase-3. LJ2 est un peptidomimétique qui combine les propriétés de la série 33c/33 h et du Δ2Me-TRP601. LJ2 inhibe préférentiellement et irréversiblement la Caspase-2 [47].
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.