")
")
| Issue |
Med Sci (Paris)
Volume 35, Number 6-7, Juin-Juillet 2019
|
|
|---|---|---|
| Page(s) | 535 - 543 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/2019093 | |
| Published online | 05 July 2019 | |
Instabilité des microsatellites et cancer
De l’instabilité du génome à la médecine personnalisée
Microsatellite instability and cancer: from genomic instability to personalized medicine
1
Sorbonne Université, UPMC Univ Paris 06, Inserm, UMRS 938, Équipe Instabilité des microsatellites et cancer, Centre de recherche Saint Antoine, 75012 Paris, France
Équipe labellisée par la Ligue nationale ontre le cancer et SIRIC CURAMUS, APHP.6
2
Sorbonne Université, Department of Digestive Surgery, AP-HP, Hôpital Saint-Antoine, F-75012, Paris, France
3
AP-HP, Service d’anatomie et cytologie pathologiques, 75012 Paris, France
4
Département d’hépatogastroentérologie, Hôpital universitaire de Poitiers, 86021 Poitiers, France
5
Département de gastroentérologie et oncologie digestive, Hôpital Européen George Pompidou, APHP, 75015 Paris, France
* This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
L’instabilité des séquences répétées du génome (appelées microsatellites) est une conséquence de l’inactivation fonctionnelle du système de réparation des erreurs produites au cours de la réplication de l’ADN (système MMR, mismatch repair). Elle signe un phénotype tumoral fréquent appelé MSI (microsatellite instable) qui a été mis en évidence il y a un peu plus de 20 ans. Les cancers MSI sont fréquents chez l’homme, associés à de nombreuses localisations primitives (côlon, estomac, endomètre, etc.). Ils peuvent être héréditaires ou, le plus souvent, de survenue sporadique. Cet article propose une synthèse des travaux dédiés à l’étude des cancers MSI menés par des chercheurs et médecins français récompensés par le prix Jean et Madeleine Schaeverbeke de la Fondation de France. Depuis 20 ans, leur activité a grandement contribué à améliorer nos connaissances sur ce mode original de tumorigenèse, jetant les bases d’une médecine personnalisée de ces tumeurs chez l’homme, en pleine émergence aujourd’hui.
Abstract
The human tumor phenotype referred to as MSI (Microsatellite Instability) is associated with inactivating alterations in MMR genes (Mismatch Repair). MSI was first observed in inherited malignancies associated with Lynch syndrome and later in sporadic colon, gastric and endometrial cancers. MSI tumors develop through a distinctive molecular pathway characterized by genetic instability in numerous microsatellite DNA repeat sequences throughout the genome. In this article, french researchers and physicians who have been recently awarded by the Fondation de France (Jean and Madeleine Schaeverbeke prize) make a sum of their activity in the MSI cancer field for more than 20 years. Their findings have greatly contributed to increase our knowledge of this original cancer model, laying the foundation for a personalized medicine of MSI tumors.
© 2019 médecine/sciences – Inserm
 Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (http://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l'utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (http://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l'utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.

Vignette (Photo @ Inserm - Julio C. Valencia).
La découverte d’un lien entre la survenue de certains cancers et l’existence d’anomalies du système de réparation des erreurs de réplication de l’ADN (système MMR pour mismatch repair) a ouvert, il y a 25 ans, de nouveaux horizons dans l’étude de la génétique des tumeurs chez l’homme [1]. Dans ces tumeurs, l’inactivation du système MMR est systématiquement observée et conduit à un phénotype d’hyperinstabilité de l’ADN à l’échelle du nucléotide. En d’autres termes, la séquence d’ADN tumoral est fréquemment remaniée par des mutations somatiques ponctuelles en nombre. Ces remaniements affectent en premier lieu des séquences répétées du génome, appelées microsatellites, avec des insertions/délétions mono ou dinucléotidiques à l’origine de cancers MSI (microsatellite instable), sans autre réarrangement génomique majeur (pas d’instabilité chromosomique ou à un taux faible, comparé à ce qui est observé dans d’autres types tumoraux) [2].
Les cancers MSI sont fréquents chez l’homme. Ils peuvent être héréditaires, intégrés dans le cadre du syndrome de Lynch, ou cancer colorectal héréditaire sans polypose (1/3 des cas environ), favorisés du fait de mutations constitutionnelles hétérozygotes d’un des gènes codant les protéines majeures du système MMR : MLH(mutL homolog human)1,2, puis MSH6 (MutS homolog human 6) et PMS2 (postmeiotic segregation increased 2) (par ordre de fréquence)1 [3]. Le syndrome de Lynch représente une des prédispositions au cancer les plus fréquentes chez l’homme. Dans ce syndrome, les patients peuvent développer différents types de tumeurs (côlon, estomac, endomètre, voies biliaires, rein, uretères, pancréas, lymphomes, leucémies, etc.), le plus souvent à un âge jeune (avant 60 ans). Une variante plus sévère de ce syndrome a été récemment rapportée. Appelé syndrome CMMRD (pour constitutive MMR-deficiency syndrome), il concerne des patients ayant des mutations germinales bi-alléliques d’un des gènes du système MMR (par ordre de fréquence : PMS2, MSH6, puis MLH1, MSH2), entraînant l’émergence de tumeurs MSI dès l’enfance avec, en particulier, des leucémies, des lymphomes, des cancers du côlon et des tumeurs cérébrales d’une particulière gravité sur un plan pronostique [4]. Les cancers MSI demeurent néanmoins dans leur majorité de survenue sporadique (dans deux tiers des cas environ), sans agrégation familiale. Au total, un phénotype MSI est rencontré dans 10 à 15 % de certaines tumeurs épithéliales (cancers colorectaux, de l’estomac, de l’endomètre), et dans une proportion faible à très faible (1 à 5 %) de nombreux autres cancers, en grande majorité des adénocarcinomes, sans d’ailleurs que nous ayons encore aujourd’hui une vision globale totalement arrêtée de leur spectre clinique (cancer de la prostate, du rein, du poumon, de l’ovaire, de la tête et du cou, du foie, de la vessie, glioblastomes, gliomes cérébraux de bas grade, cancer du sein, etc.) [5].
Dans cette revue, nous présentons une synthèse des travaux dédiés à l’étude des cancers MSI. Ces travaux ont été menés par des chercheurs et médecins qui ont consacré toute ou une partie de leur activité à ces tumeurs durant ces 20 dernières années. Ils ont trait à la mise au point de méthodes diagnostiques ayant permis d’identifier ce phénotype tumoral chez l’homme, à la mise en évidence de facteurs de risque favorisant leur émergence dans la population, à la caractérisation de mécanismes physiopathologiques sous-jacents impliqués dans l’initiation et la progression tumorale MSI, à l’identification de marqueurs d’intérêt pronostique ou même théranostique dans ces cancers, ou encore à l’étude de l’impact des MSI sur la réponse à la chimiothérapie (Figure 1). Ces travaux, résumés ici2, donnent un aperçu de la recherche nationale sur ce type tumoral. Ils ont été soutenus par la Fondation de France qui a récompensé chacun de ces chercheurs en leur attribuant le prix Jean et Madeleine Schaeverbeke.
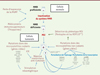 |
Figure 1. Étapes de la tumorigenèse MSI. MSI : instabilité des microsatellites ; ICK : intestinal cell kinase. |
Diagnostic du phénotype MSI dans les tumeurs
Le phénotype MSI d’une tumeur est aujourd’hui très fréquemment recherché chez les patients présentant un cancer colorectal (CCR)3. Cet examen est de plus en plus recommandé pour d’autres types tumoraux (voir plus haut, le spectre des tumeurs MSI). L’intérêt de la mise en évidence d’un tel phénotype dans un cancer est double. Il permet d’une part d’identifier, ou de contribuer à identifier, les patients à risque pour le syndrome de Lynch et le CMMRD (tumeurs MSI héréditaires). Il représente d’autre part, et notamment à un stade métastatique, un critère d’orientation thérapeutique puisque les patients présentant un cancer MSI métastatique sont désormais candidats pour être traités par de nouvelles thérapies (blocage des points de contrôle immunitaire par immunothérapie par anticorps anti-PD-1 [programmed death-1], anti-CTLA-4 [cytotoxic T-lymphocyte associated protein 4], ou autres ; voir ci-dessous pour plus de détails) [6]. Certains auteurs de cette revue ont contribué à la mise au point de méthodes de référence utilisées aujourd’hui en routine à l’international pour identifier le statut MSI à partir de l’ADN tumoral. Il s’agit en particulier du test Pentaplex® qui repose sur la co-amplification de 5 marqueurs microsatellites dans l’ADN tumoral et permet d’identifier de manière simple, sensible et spécifique, le phénotype MSI d’une tumeur : ces marqueurs sont en effet très stables dans les ADN normaux et quasi-systématiquement instables dans les ADN tumoraux de cellules déficientes en MMR [7, 8]. Ces méthodes ont fait l’objet de simplifications ultérieures [9]. L’avenir est à leur potentialisation afin de permettre la détection du phénotype MSI directement à partir du sang des patients (détection d’ADN tumoral circulant sans nécessité d’un accès direct à la tumeur), ce qui présente un intérêt en cas de patients métastatiques non opérables ou pour le suivi de la maladie résiduelle après traitement. Cette détection dans le sang périphérique a également un intérêt pour l’évaluation de MSI dans les tissus sains de patients prédisposés à ce type de cancer (patients atteints de syndrome de Lynch ou de CMMRD) puisqu’il a été rapporté que ces malades pouvaient présenter un bruit de fond d’instabilité microsatellitaire dans certains tissus, avant même la survenue de cancer, chez eux ou leurs apparentés [10].
Aujourd’hui, les CCR sont systématiquement examinés4 pour le statut MSI à partir d’une analyse de l’ADN tumoral par ces méthodes de référence. Plusieurs études ont exploité ces méthodes afin d’identifier en particulier les patients souffrant de syndrome de Lynch, prédisposés à la survenue de CCR MSI héréditaires. Une série de 1 040 cas de CCR réséqués entre 2005 et 2009 a ainsi pu être analysée. Cent-cinq tumeurs présentaient une perte d’expression protéique du système MMR et/ou une instabilité microsatellitaire. Plus d’un tiers des syndromes de Lynch confirmés (avec mise en évidence d’une mutation germinale MMR) ne présentaient aucun signe clinique évocateur d’une prédisposition familiale et n’auraient donc pas été identifiés sans cette recherche systématique du phénotype MSI sur l’ADN tumoral [11]. Cette très large étude montre ainsi qu’il est crucial de proposer systématiquement un examen du statut MSI sur l’ADN tumoral chez les patients présentant un CCR afin de repérer ceux qui, parmi eux, sont atteints de syndrome de Lynch et donc prédisposés au développement de néoplasies MSI familiales.
Identification de contextes cliniques et de facteurs de risques favorisant la déficience MMR
En dehors d’une prédisposition héréditaire liée à l’existence d’une mutation germinale MMR hétérozygote (syndrome de Lynch) ou homozygote (CMMRD), l’émergence de tumeurs MSI peut être favorisée dans certains contextes cliniques, par des mécanismes bien spécifiques. Nous avons pu en effet montrer la survenue de tumeurs intestinales MSI ou de lymphomes MSI chez des patients souffrant de maladies inflammatoires chroniques de l’intestin (MICI)5 [12, 13]. On estime actuellement que 100 000 personnes en sont atteintes en France (60 000 pour la MC, 40 000 pour la RCH). Ces malades présentent un risque accru de développer un cancer colorectal comparés à la population générale et le phénotype MSI est retrouvé dans une fraction significative des tumeurs colorectales compliquant une MICI (avec une fréquence globale de 8,3 %, dont 7,8 % chez les patients RCH et 9,6 % chez les patients atteints de MC) [12]. Ces tumeurs intestinales MSI qui surviennent dans un contexte de MICI, ont des caractéristiques qui ressemblent à celles des tumeurs MSI héréditaires (c’est-à-dire une survenue chez l’adulte jeune, une perte d’expression d’une des 4 protéines MMR : MLH1, MSH2, MSH6 ou PMS2 ; les cas sporadiques sont essentiellement liés à la perte d’expression de MLH1 dans la tumeur par l' hyperméthylation de son promoteur) [14].
D’autres travaux connexes ont permis d’identifier la survenue de lymphomes MSI dans un contexte de greffe d’organe, c’est-à-dire chez des patients ayant bénéficié d’un don d’organe et ayant été ensuite traités par immunosuppresseurs au long cours (thérapie anti-rejet dans un contexte post-greffe) [15]. Contrairement aux lympho-proliférations liées à la résurgence du virus d’Epstein-Barr (EBV), qui sont précocement observées après la greffe, les lymphomes MSI sont négatifs pour le virus, et ils surviennent en moyenne plus de 5 ans après la greffe d’organe [16].
Sur un plan mécanistique, l’inactivation du MMR dans ces situations particulières est favorisée du fait de processus génotoxiques. En effet, l’inflammation chronique (dans le cas des MICI) ou la prise de certains médicaments (comme les thiopurines : l’azathioprine [ImurelTM]) prescrits dans les MICI mais aussi chez les patients greffés d’organes, induisent des adduits6 de l’ADN, en particulier la modification de guanines [17]. Ce processus conduit dans les tissus des patients (les cryptes coliques, le tissu lymphoïde, etc.) à la génération de mésappariements de bases, normalement reconnus par le système MMR mais qui ne les répare pas efficacement. Les cellules accumulent ainsi des lésions sur leur l’ADN qui conduisent à un arrêt du cycle cellulaire puis à la mort des cellules par apoptose. C’est dans ce contexte que l’inactivation du système MMR représente un mécanisme d’échappement à la mort cellulaire, conférant aux cellules dans lesquelles il survient un avantage sélectif en culture, ce qui favorise leur transformation via des processus découlant de l’instabilité microsatellitaire. Ce scénario a été validé, en particulier dans un modèle animal par l’administration de thiopurines (ImurelTM) à des souris, démontrant ainsi que ce type de molécule contribuait à favoriser l’émergence de tumeurs MSI [14, 18].
Caractérisation des événements somatiques conduisant à la transformation des cellules déficientes en MMR
L’inactivation du système MMR ne constitue pas en tant que tel un événement transformant. C’est l’accumulation dans l’ADN tumoral de mutations somatiques engendrées par le phénotype MSI qui participe au processus de transformation multi-étapes conduisant à générer une tumeur [2]. Dans les tumeurs MSI, l’instabilité génétique est élevée (environ une centaine de mutations par mégabase en moyenne), à l’origine de nombreux évènements somatiques, principalement dans les microsatellites. Ces altérations somatiques microsatellitaires sont susceptibles de participer au développement tumoral lorsqu’elles modifient la fonction de gènes ayant un rôle dans l’oncogenèse [19, 20]. Il convient de noter également que les tumeurs MSI accumulent des mutations somatiques récurrentes au niveau de séquences génomiques non répétées, en particulier sur quelques gènes majeurs du processus transformant (BRAF, KRAS, PIK3CA, APC, CTNNB1 7, etc.) [21].
Un répertoire des altérations liées au MSI au niveau de répétitions microsatellitaires codantes (notion de gènes cibles de MSI) a pu être dressé [22]. Ces mutations affectent de manière tissu-spécifique (le répertoire de mutations varie selon les cancers MSI du côlon, de l’estomac, de l’endomètre) des gènes impliqués dans des processus biologiques divers, comme la régulation du cycle et/ou de la prolifération cellulaire (TGFBR2, IGF2R, TCF4, AXIN2, PTEN, RIZ, etc.), la régulation de l’apoptose (BAX, CASP5, BCL10, APAF1, FAS, etc.), de la réparation et la signalisation des dommages de l’ADN (RAD50, BLM, MRE11, MSH3, MSH6, MBD4, MLH3, CHK1, ATR, etc.) (pour revue voir [20]). L’instabilité des microsatellites altère également des dizaines d’autres gènes cibles dont le rôle n’est pas forcément pertinent dans ces cancers (pas de rôle dans la transformation maligne). Dans la plupart des cas, les conséquences fonctionnelles de ces mutations sont encore mal définies. Les travaux de génomique dédiés au CCR MSI et l’analyse des fréquences mutationnelles de ces gènes dans l’ADN tumoral ont montré que seule une fraction minoritaire de ces mutations présente une fréquence mutationnelle importante au cours du développement du cancer et peut en conséquence représenter les gènes majeurs du développement tumoral MSI [23, 24]. À l’heure actuelle, grâce à l’utilisation du séquençage à haut débit, la recherche sur l’instabilité microsatellitaire progresse pour la caractérisation de ces répertoires de mutations [25]. Certains des auteurs de cette revue ont été impliqués dans la description fine de l’exome d’une large série de CCR MSI. Ce travail, qui a donné lieu à la publication de la première analyse génomique à haut débit des CCR MMR-déficients [26], a permis d’identifier les mutations dont la fréquence émerge d’un bruit de fond d’instabilité très important caractérisant ce type tumoral. Ces mutations somatiques de séquences répétées ou non jouent probablement un rôle clé dans ces cancers (sélection positive). Ce travail a montré également que la tumorigenèse MSI est un processus complexe qui engendre aussi des altérations somatiques en nombre, dont la fréquence s’est révélée être moins élevée qu’attendue (après prise en compte de la taille et de la nature de ces répétitions à chaque fois dans le modèle) du fait de leur caractère probablement délétère (sélection négative).
Découverte de la mutation d’HSP110 dans les CCR MSI et caractérisation de son impact physiopathologique et clinique
Les protéines chaperonnes, ou HSP (heat shock protein), interviennent dans de très nombreux processus biologiques. Comme leur nom l’indique, elles stabilisent les protéines en les chaperonnant, facilitant ainsi leur repliement dans différentes conditions, les protégeant contre des phénomènes d’agrégation protéique, ou favorisant leur catabolisme lorsqu’il s’agit de protéines endommagées ou dégradées. L’expression des HSP est induite par des stress divers (thermique, stress oxydant, agressions chimiques, irradiations, etc.). Chez les mammifères, il existe plusieurs familles d’HSP, classées selon leur masse moléculaire. Dans les cancers, le rôle des chaperonnes est pléiotrope. Elles sont très souvent surexprimées et jouent un rôle oncogénique en permettant aux cellules malignes de s’adapter à différentes conditions de stress, endogènes comme exogènes, au cours du développement tumoral, y compris pendant la phase de traitement par chimiothérapie (facteur de chimiorésistance) [27]. Les HSP participent activement à la transformation tumorale par de nombreux mécanismes, et jouent aussi un rôle dans l’immunité anti-tumorale, en étant en particulier sécrétées dans le milieu extra- cellulaire [28].
En 2011, nous avons rapporté un fait assez surprenant concernant l’une de ces protéines chaperonnes dans les cancers colorectaux (CCR) [29], avec l’identification d’une mutation d’un gène codant une HSP spécifiquement dans les CCR de phénotype MSI. Cette mutation représentait ainsi le premier exemple d’altération d’une chaperonne dans un cancer chez l’homme. Elle touche le gène HSP110 (encore appelé HSPH1), au niveau d’un microsatellite, T17, intronique et localisé dans une séquence polypyrimidiques en amont du site accepteur d’épissage de l’exon 9 (intron 8). Ce microsatellite est muté dans les ADN tumoraux de CCR MSI par la délétion de 1 à 6 bases. Ces délétions alléliques du microsatellite T17 entraîne, lorsqu’elles sont de grandes tailles (supérieures à 5 paires de bases, ou délétions larges), un épissage aberrant de l’exon 9 par saut d’exon et sont donc à l’origine de la synthèse d’une protéine tronquée (mutation frameshift, création d’un codon STOP prématuré). Environ 30 % des CCR MSI présentent de telles délétions larges de manière bi-allélique dans l’ADN tumoral. Dans ces tumeurs, la protéine HSP110 sauvage n’est de fait plus exprimée, ou à des niveaux très bas. Cette inactivation somatique d’HSP110 a des conséquences majeures sur un plan fonctionnel. En effet, les cellules nullizygotes pour HSP110 (mutées sur les deux allèles) deviennent très sensibles au stress, comme un stress apoptotique, et sont ainsi hyper-sensibles à des traitements par différents agents, incluant ceux utilisés dans les protocoles de chimiothérapie (5-Fluorouracile et Oxaliplatine) [29]. Cliniquement, nous avons pu établir, dans le cadre d’analyses de cohortes de patients en provenance de différents centres hospitaliers, en France et à l’étranger, que les malades présentant un CCR n’exprimant plus significativement HSP110 (tumeur HSP110 large/large) étaient d’excellents répondeurs à la chimiothérapie par 5-Fluorouracile utilisé seul et par FOLFOX (une combinaison de 5-Fluorouracile et d’Oxaliplatine) [30].
Ces résultats sont intrigants puisqu’ils révèlent de manière assez paradoxale que la mutation d’une protéine chaperonne, fréquente dans les CCR MSI, générée par une instabilité microsatellitaire somatique de l’ADN, est clairement délétère pour les cellules tumorales et diminue leur capacité de résistance aux drogues. Ils suggèrent le scénario selon lequel certaines mutations somatiques ne peuvent être évitées par les cellules tumorales MSI au niveau de grands microsatellites comme le T17 d’HSP110 qui constituent des points obligatoires de mutations dans ce modèle tumoral hyper-instable. Des cribles (analyse de l’exome tumoral, profils de séquençage d’ARN [RNA-seq]) sont en cours, à la recherche d’autres mutations délétères dont la survenue serait observée dans les mêmes conditions, en particulier des mutations touchant les processus d’épissage. Des travaux plus récents nous ont également permis de mettre en évidence d’autres effets délétères de la mutation d’HSP110 dans les CCR MSI au cours du développement tumoral [31].
Impacts pronostique et prédictif de réponse à la chimiothérapie du MSI
L’amélioration du pronostic conféré par le statut MSI chez les patients atteints de cancer colorectal a été évoqué dès les premières descriptions de ce type d'instabilité génétique. Par la suite, de nombreuses études, rétrospectives pour la plupart, incluant des patients avec un CCR de stade 2 ou 3 (tumeurs respectivement avec ou sans envahissement ganglionnaire mais non métastatiques à d’autres organes) [32], ou des patients présentant un CCR tous stades confondus [33], ont également rapporté des survies prolongées en rapport avec le statut MSI des tumeurs. Une méta-analyse fondée sur 31 études ayant inclus au total 7 642 patients tous stades confondus, dont 1 277 MSI, a montré une réduction de 35 % du risque de décès chez les patients MSI par rapport aux patients ne présentant pas d’instabilité des microsatellites (MSS, stabilité des microsatellites) [34]. Le bénéfice de survie observé pour les patients MSI persistait lorsque l’analyse était restreinte à ceux atteints d’une tumeur de stade 2 ou 3.
Depuis la publication de l'essai MOSAIC8, l’utilisation de l’oxaliplatine en combinaison avec le 5-Fluorouracile constitue le traitement adjuvant de référence après la résection des CCR de stade 3 [35]. En revanche, l’indication d’une chimiothérapie adjuvante pour les tumeurs de stade 2 reste moins bien standardisée et limitée aux patients présentant des facteurs de haut risque de récidive tumorale (stade T4 : présence d’une perforation tumorale, etc.). Les modèles précliniques réalisés avec des lignées cellulaires ont suggéré que les tumeurs MSI étaient associées à une résistance au 5-Fluorouracile. Plusieurs études cliniques ont évalué le rôle du MMR dans la réponse au traitement adjuvant par 5-Fluorouracile chez les patients opérés pour un CCR de stade 2 ou 3. Des résultats contradictoires ont été obtenus, rapportant pour certains un bénéfice du 5-Fluorouracile pour les tumeurs MSI [36], tandis que d’autres concluaient à l’absence d’efficacité, voire à un effet potentiellement délétère pour les tumeurs MSI de stade 2 [32]. Dans une étude publiée en 2010, Sargent et al. ont évalué l’influence du phénotype MSI à partir d'une série de 457 patients inclus dans cinq essais randomisés évaluant le traitement adjuvant par 5-Fluorouracile versus une chirurgie seule se résumant à la résection d’un cancer du côlon de stade 2 et 3. Dans cette analyse, le phénotype MSI versus MSS s’est révélé être un facteur prédictif de résistance au 5-Fluorouracile, quel que soit le stade de la tumeur. Ces données ont été combinées à celles obtenues antérieurement par Ribic et al., permettant d’augmenter la série à 1 027 patients traités pour un cancer du côlon de stade 2 et 3 [32]. Cette analyse a confirmé le bon pronostic du statut MSI ainsi que l’absence d’efficacité de la chimiothérapie adjuvante par 5-Fluorouracile pour les tumeurs MSI de stade 3, voire un effet délétère pour les tumeurs MSI de stade 2. Les premières études rétrospectives, dont les nôtres [37], avaient montré que l’addition de l’oxaliplatine au 5-Fluorouracile pouvait restaurer l’efficacité de la chimiothérapie adjuvante pour les tumeurs MSI de stade 3. Ces résultats ont ensuite été confirmé sur une large cohorte de 433 patients MSI de stade 2 ou 3 traités par chirurgie seule ou associée à une chimiothérapie adjuvante par fluoropyrimidine avec ou sans oxaliplatine [38]. L’utilisation d’une fluoropyrimidine seule n’améliore pas la survie des patients par rapport au traitement chirurgical. En revanche, l’adjonction de l’oxaliplatine à une fluoropyrimidine permet une amélioration significative de la survie des patients présentant une tumeur de stade 3, mais pas de ceux ayant une tumeur de stade 2. Dans le cadre d’une étude rétrospective multicentrique de 521 patients avec un CCR MSI non métastatique, 3 facteurs prédictifs de la survie sans récidive (SSR) ont été définis : l’occlusion intestinale, la présence d’emboles vasculaires et un stade T4 [39], orientant la décision et les modalités de la chimiothérapie adjuvante dans ce type de cancer (Figure 2)
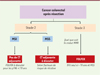 |
Figure 2. Algorithme pour la décision thérapeutique de chimiothérapie adjuvante dans les CCR de stade 2-3. CT : chimiothérapie. |
Des analyses rétrospectives réalisées sur des échantillons tumoraux de 1 008 patients inclus dans l’étude MOSAIC, ont, par la suite, montré que l’adjonction de l’oxaliplatine au 5-Fluorouracile apportait un bénéfice de survie à la fois pour les tumeurs MSI et MSS. Plus récemment, une étude regroupant deux essais randomisés ayant inclus 4 674 patients traités par FOLFOX avec ou sans cétuximab (un anticorps monoclonal bloquant le récepteur du facteur de croissance épidermique, EGFR) pour une tumeur de stade 3, a montré que le phénotype MSI, versus MSS, était un facteur de bon pronostic, et cela quelle que soit l’origine de l'inactivation du système MMR (sporadique ou familiale).
D’autres études cliniques sont aujourd’hui en cours pour mieux définir l’intérêt clinique de différents biomarqueurs dans les CCR MSI (identification de mutations ou analyse d’expression en immunohistochimie)9. Des protocoles de recherche clinique sont également élaborés afin d’étudier la problématique des CCR MSI de stade 4 (stade métastatique). Le stade métastatique est rarement atteint dans les CCR MSI (moins de 5 % des cas de CCR métastatiques), mais il représente un stade particulièrement grave de la maladie (pronostic plus sombre que celui des CCR MSS au même stade) [40]. Les analyses de cohortes10 en cours semblent indiquer que le pronostic et la chimiosensibilité des CCR MSI métastatiques seraient différents de ceux des CCR MSS métastatiques, mais ces résultats doivent être confirmés par des études prospectives.
Récemment, un essai de phase 2 réalisé chez des patients avec une tumeur métastatique a montré que le statut MSI était un facteur prédictif de réponse à l’immunothérapie [6]. Le statut MSI/MSS de tumeurs isolées de patients présentant un CCR métastatique, inclus dans un essai clinique multicentrique afin de bénéficier d’un traitement par immunothérapie, a été contrôlé [41]. Des erreurs diagnostiques (diagnostic erroné à l’inclusion par les centres investigateurs) sur le statut MSI de tumeurs qui étaient en fait MSS et avaient donc été incluses à tort au sein de cette cohorte [42], ont été identifiées. La mise en évidence de ces erreurs diagnostiques a permis, en particulier, d’expliquer l’absence de réponse au traitement observée chez la majorité des patients hyperprogresseurs, venant donc constituer la première cause de « résistance » à l’immunothérapie dans les CCR MSI métastatiques. Ces résultats devront être vérifiés sur d’autres cohortes à l’avenir.
Le phénotype MSI dans les CCR est donc un marqueur moléculaire de bon pronostic, qui semble également prédire l'absence d'efficacité de la chimiothérapie adjuvante par le 5-Fluorouracile seul. Les récentes données cliniques suggèrent que l'adjonction de l'oxaliplatine à un traitement par le 5-Fluorouracile pourrait rétablir le bénéfice de la chimiothérapie adjuvante pour les patients avec un cancer du côlon MSI de stade 3. Sur la base de ces données, les recommandations nationales préconisent une chimiothérapie adjuvante par fluoropyrimdine plus oxaliplatine pour les patients opérés d’un cancer du côlon de stade 3 quel que soit le phénotype MMR (MSI ou MSS). Pour les patients avec un cancer du côlon de stade 2 présentant des facteurs de haut risque de récidive, ceux dont la tumeur est MSS pourraient être traités par fluoropyrimidine seule ou associée à l’oxaliplatine. En revanche, l’indication d’une chimiothérapie adjuvante n’est pas retenue pour les tumeurs MSI de stade 2, compte-tenu du bon pronostic et de l’inefficacité du 5-Fluorouracile dans ce contexte. Une étude actuellement en cours11 évalue l’intérêt d’associer l’atezolimumab (un anticorps monoclonal dirigé contre le ligand de PD-1, PD-L1) avec le FOLFOX après la résection d’un cancer du côlon MSI de stade 2 (Tableau I).
Caractéristiques pronostiques et de réponse à la chimiothérapie des CCR (MSI versus MSS). MSI : instabilité des microsatellites ; MSS : stabilité des microsatellites ; CT : chimiothérapie ; anti-ICK : anti-immunocheckpoint.
Étude de l’infiltrat immunitaire des cancers colorectaux MSI
Dans les tumeurs MSI et le cancer colorectal en particulier, l’instabilité microsatellitaire produit un nombre important de protéines mutantes immunogènes. En effet, les insertions/délétions typiquement générées du fait du statut MSI au niveau des centaines de microsatellites codants sont à l’origine d’autant de décalages de lecture (mutation frameshift) sur les transcrits des gènes cibles correspondants. Les protéines mutantes ainsi traduites possèdent en conséquence des extrémités C-terminales aberrantes néo-antigéniques qui sont reconnues comme telles par le système immunitaire du patient [43]. L’expression de ces mutants est néanmoins régulée et il existe, en particulier, des mécanismes de catabolisme dans la cellule qui identifient et dégradent les ARN messager comportant un codon STOP prématuré, comme ceux générés en nombre dans la cellule tumorale déficiente en MMR du fait de MSI. Ce système, appelé NMD (nonsense mediated-mRNA decay), a une activité importante dans la cellule tumorale MSI. Il permet la diminution de l’expression de nombreux mutants favorisés par MSI dans la tumeur avec des conséquences à la fois sur la croissance tumorale et la réponse immunitaire des patients. Il semble en particulier que plus l’expression des facteurs du NMD est importante, plus celle des néoantigènes produits par la cellule tumorale est diminuée, rendant la tumeur moins visible par le système immunitaire (avec, en particulier, la diminution du nombre de lymphocytes T CD3 infiltrants) [23, 24, 44].
Les CCR MSI présentent généralement un infiltrat lymphocytaire T important, par rapport à ce qui est rencontré dans les CCR sans instabilité des microsatellites (MSS). Nous avons montré que l’infiltrat T (lymphocytes CD3+) de ces tumeurs est associé au nombre de mutations mais aussi à certaines mutations des gènes cibles du MSI. Ces résultats suggèrent que plus le nombre de mutations est important, plus il existe de lymphocytes T spécifiques, mais aussi qu’il existe des mécanismes d’échappement à cette réponse immunitaire puisque le nombre de mutations est également corrélé à la progression tumorale. Afin de caractériser plus précisément les néo-antigènes immunogènes, nous avons analysé la relation entre mutations cibles de l’instabilité microsatellitaire et infiltrats T CD8+ (lymphocytes T cytotoxiques) et Foxp3+ (lymphocytes T régulateurs, LTreg) [45]. L’infiltrat T cytotoxique augmente significativement avec le nombre de mutations ; il est associé à des mutations qui décalent le cadre de lecture de certains gènes (ASTE1, HNF1A, et TCF4), suggérant l’établissement d’une réponse immunitaire spécifique vis-à-vis des néo-antigènes produits à la suite de ces mutations.
Il nous a paru important de connaître l’impact pronostique de l’infiltration lymphocytaire dans ces tumeurs, notamment les lymphocytes T cytotoxiques et régulateurs. Un faible infiltrat de LTreg Foxp3+ est associé à un stade avancé et à des critères d’invasion métastatique précoce. Ces résultats surprenants suggèrent que l’infiltrat T régulateurs, habituellement considéré comme immunosuppresseur, est associé à une moindre invasivité tumorale dans les CCR MSI. On peut penser que plus la réponse immunitaire est importante, plus les LTreg sont nombreux et meilleur est le pronostic. Néanmoins, il est important de mieux caractériser la réponse immunitaire dans ces tumeurs, ainsi que leurs mécanismes d’échappement. Dans un travail collaboratif, nous avons caractérisé le profil immunitaire de ces tumeurs [46]. Les CCR MSI ont un « immunoscore » élevé avec une forte réponse lymphocytaire de type Th1, des taux importants de lymphocytes T mémoires mais également une forte expression de molécules inhibitrices de la réponse immune, comme PD-1 et PD-L1. L’interaction entre PD-1 et PD-L1 entraîne un épuisement fonctionnel des lymphocytes T activés et représente un mécanisme d’échappement tumoral à la réponse immunitaire des CCR MSI. Des essais thérapeutiques récents ont montré que ces tumeurs sont très sensibles aux inhibiteurs de points de contrôle immunitaire (immune checkpoint inhibitors) [6]. Toutes ces données générées sur l’immunité anti-tumorale des CCR MSI seront bientôt validées par des études ancillaires adossées aux essais thérapeutiques d’inhibiteurs de points de contrôle immunitaire, afin de mieux comprendre l’efficacité particulière de ces traitements dans les tumeurs MSI. Cela est déjà prévu dans le cadre d’un essai thérapeutique coordonné par la FFCD (Fédération francophone de cancérologie digestive) avec un anticorps anti-PD-1 en 2e ligne, dans les CCR MSI métastatiques.
Aujourd’hui, des analyses à haut débit de l’infiltrat immunitaire des tumeurs sont développées avec des approches de séquençage d’ARN (RNA-seq) et de transcriptomique. Dans une étude récente qui porte sur plus de 1 000 CCR, incluant 260 CCR MSI, en provenance de 2 séries multi-centriques (1 cohorte en provenance du TCGA, 1 cohorte française, série CIT), nous avons montré que c’est l’expression des points de contrôle immunitaire (PD-1, PD-L1, IDO1, TIM3, LAG3) qui influence majoritairement et négativement le pronostic dans les CCR MSI par rapport à d’autres marqueurs immunitaires, dont ceux de l’immunoscore en particulier [47]. Dans les CCR MSS, l’inverse se produit, avec un impact significatif de l’expression des marqueurs de l’immunoscore (CD3 et CD8, en particulier), comme cela a déjà été montré, et un impact mineur des points de contrôle. Ainsi, chez les patients présentant un CCR, le pronostic de la maladie est déterminé par une balance entre l’expression des marqueurs de l’immunoscore et celle des points de contrôle, avec un impact clinique important de l’une ou de l’autre de ces composantes antagonistes de la réponse immunitaire.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Glossaire
Glossaire
APAF1 : : apoptotic peptidase-activating factor 1
APC : : adenomatous polyposis coli - APC regulator of WNT signaling pathway
ASTE1 : : asteroid homolog 1
ATR : : ATR serine/threonine kinase
AXIN2 : : axin-related protein 2 or axis inhibition protein 2
BAX : : BCL2-associated X
BCL10 : : BCL10 immune signaling adaptor
BLM : : BLM RecQ like helicase
BRAF : : B-Raf proto-oncogene, serine/threonine kinase
CASP5 : : caspase 5
CHK1 : : checkpoint kinase 1
CMMRD : : constitutional mismatch repair deficiency syndrome
CTLA-4 : : cytotoxic T-lymphocyte-associated protein 4
CTNNB1 : : catenin beta 1
EGFR : : epidermal growth factor receptor
FAS : : Fas cell surface death receptor
FOXP3 : : forkhead box P3
HNF1A : : HNF1 homeobox A
IDO1 : : indoleamine 2,3-dioxygenase 1
IGF2R : : insulin like growth factor 2 receptor
KRAS : : KRAS proto-oncogene, GTPase
LAG3 : : lymphocyte activating 3
MBD4 : : methyl-CpG binding domain 4, DNA glycosylase
MGMT : : O6-methylguanine-DNA methyltransferase
MLH1 : : mutL homolog 1
MLH3 : : mutL homolog 3
MMR : : mismatch repair system
MRE11 : : MRE11 homolog, double-strand break repair nuclease
MSH2 : : mutS homolog 2
MSH3 : : mutS homolog 3
MSH6 : : mutS homolog 6
NMD : : nonsense-mediated mRNA decay
PD-1 : : programmed cell death 1
PD-L1 : : programmed cell death 1 ligand 1
PIK3CA : : phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha
PMS2 : : PMS1 homolog 2, mismatch repair system component
PTEN : : phosphatase and tensin homolog
RAD50 : : RAD50 double strand break repair gene
RIZ : : PR/SET domain 2 (PRDM2)
TCF4 : : transcription factor 4
TGFBR2 : : transforming growth factor beta receptor 2
TIM3 ou HAVCR2 : : hepatitis A virus cellular receptor 2
Le systè me MMR est composé de 4 gè nes appelé s MLH1, MSH2, MSH6 et PMS2. Les proté ines codé es par ces gènes interagissent pour identifier puis corriger les mé sappariements de l’ADN qui ré sultent d’erreurs commises par l’ADN polymé rase lors de la ré plication de l’ADN.
Les références correspondantes sont en bleu dans la liste bibliographique.
La recherche de MSI systématique est réalisée si l’âge du patient est inférieur à 60 ans et/ou en cas d’antécédents personnels ou familiaux de cancer.
Notamment à l’hôpital Saint-Antoine qui accueille la majorité des Lauréats du Prix Jean et Madeleine Schaeverbeke signataires de cette publication.
Les MICI (maladies inflammatoires chroniques de l’intestin) regroupent la maladie de Crohn (MC) et la rectocolite hémorragique (RCH).
Produit d'une réaction d'addition entre deux unités moléculaires distinctes.
Voir Glossaire.
L'essai MOSAIC avait été conçu pour évaluer les effets du FOLFOX dans le traitement adjuvant des cancers coliques de stade 2 et 3.
Étude COLOMIN : Cohorte nationale de cancers colorectaux avec instabilité microsatellitaire.
Cohorte COLOMIN META.
Étude NCT02912559.
Références
- Aaltonen LA, Peltomaki P, Leach FS, et al. Clues to the pathogenesis of familial colorectal cancer. Science 1993 ; 260 : 812–816. [Google Scholar]
- Duval A, Hamelin R. Mutations at coding repeat sequences in mismatch repair-deficient human cancers: toward a new concept of target genes for instability. Cancer Res 2002 ; 62 : 2447–2454. [Google Scholar]
- Peltomaki P, Vasen H. Mutations associated with HNPCC predisposition: update of ICG-HNPCC/INSiGHT mutation database. Dis Markers 2004 ; 20 : 269–276. [CrossRef] [PubMed] [Google Scholar]
- Colas C, Coulet F, Svrcek M, et al. Lynch or not Lynch? Is that always a question?. Adv Cancer Res 2012 ; 113 : 121–166. [CrossRef] [PubMed] [Google Scholar]
- Hause RJ, Pritchard CC, Shendure J, Salipante SJ. Classification and characterization of microsatellite instability across 18 cancer types. Nat Med 2016 ; 22 : 1342–1350. [CrossRef] [PubMed] [Google Scholar]
- Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 2015 ; 372 : 2509–2520. [Google Scholar]
- Suraweera N, Duval A, Reperant M, et al. Evaluation of tumor microsatellite instability using five quasimonomorphic mononucleotide repeats and pentaplex PCR. Gastroenterology 2002 ; 123 : 1804–1811. [CrossRef] [PubMed] [Google Scholar]
- Buhard O, Suraweera N, Lectard A, et al. Quasimonomorphic mononucleotide repeats for high-level microsatellite instability analysis. Dis Markers 2004 ; 20 : 251–257. [CrossRef] [PubMed] [Google Scholar]
- Buhard O, Lagrange A, Guilloux A, et al. HSP110 T17 simplifies and improves the microsatellite instability testing in patients with colorectal cancer. J Med Genet 2016 ; 53 : 377–384. [CrossRef] [PubMed] [Google Scholar]
- Bodo S, Colas C, Buhard O, et al. Diagnosis of constitutional mismatch repair-deficiency syndrome based on microsatellite instability and lymphocyte tolerance to methylating agents. Gastroenterology 2015 ; 149 : 1017–29 e3. [Google Scholar]
- Canard G, Lefevre JH, Colas C, et al. Screening for Lynch syndrome in colorectal cancer: are we doing enough?. Ann Surg Oncol 2012 ; 19 : 809–816. [CrossRef] [PubMed] [Google Scholar]
- Svrcek M, El-Bchiri J, Chalastanis A, et al. Specific clinical and biological features characterize inflammatory bowel disease associated colorectal cancers showing microsatellite instability. J Clin Oncol 2007 ; 25 : 4231–4238. [CrossRef] [PubMed] [Google Scholar]
- Svrcek M, Buhard O, Colas C, et al. Methylation tolerance due to an O6-methylguanine DNA methyltransferase (MGMT) field defect in the colonic mucosa: an initiating step in the development of mismatch repair-deficient colorectal cancers. Gut 2010 ; 59 : 1516–1526. [CrossRef] [PubMed] [Google Scholar]
- Chalastanis A, Penard-Lacronique V, Svrcek M, et al. Azathioprine-induced carcinogenesis in mice according to Msh2 genotype. J Natl Cancer Inst 2010 ; 102 : 1731–1740. [CrossRef] [PubMed] [Google Scholar]
- Duval A, Raphael M, Brennetot C, et al. The mutator pathway is a feature of immunodeficiency-related lymphomas. Proc Natl Acad Sci USA 2004 ; 101 : 5002–5007. [CrossRef] [Google Scholar]
- Borie C, Colas C, Dartigues P, et al. The mechanisms underlying MMR deficiency in immunodeficiency-related non-Hodgkin lymphomas are different from those in other sporadic microsatellite instable neoplasms. Int J Cancer 2009 ; 125 : 2360–2366. [CrossRef] [PubMed] [Google Scholar]
- Beaugerie L, Svrcek M, Seksik P, et al. Risk of colorectal high-grade dysplasia and cancer in a prospective observational cohort of patients with inflammatory bowel disease. Gastroenterology 2013; 145 : 166–75e8. [CrossRef] [PubMed] [Google Scholar]
- Bodo S, Svrcek M, Sourrouille I, et al. Azathioprine induction of tumors with microsatellite instability: risk evaluation using a mouse model. Oncotarget 2015 ; 6 : 24969–24977. [CrossRef] [PubMed] [Google Scholar]
- Duval A, Reperant M, Compoint A, et al. Target gene mutation profile differs between gastrointestinal and endometrial tumors with mismatch repair deficiency. Cancer Res 2002 ; 62 : 1609–1612. [Google Scholar]
- Hamelin R, Chalastanis A, Colas C, et al. Clinical and molecular consequences of microsatellite instability in human cancers. Bull Cancer 2008 ; 95 : 121–132. [PubMed] [Google Scholar]
- Oliveira C, Pinto M, Duval A, et al. BRAF mutations characterize colon but not gastric cancer with mismatch repair deficiency. Oncogene 2003 ; 22 : 9192–9196. [Google Scholar]
- Duval A, Iacopetta B, Thorstensen L, et al. Gender difference for mismatch repair deficiency in human colorectal cancer. Gastroenterology 2001 ; 121 : 1026–1027. [CrossRef] [PubMed] [Google Scholar]
- El-Bchiri J, Buhard O, Penard-Lacronique V, et al. Differential nonsense mediated decay of mutated mRNAs in mismatch repair deficient colorectal cancers. Hum Mol Genet 2005 ; 14 : 2435–2442. [CrossRef] [PubMed] [Google Scholar]
- El-Bchiri J, Guilloux A, Dartigues P, et al. Nonsense-mediated mRNA decay impacts MSI-driven carcinogenesis and anti-tumor immunity in colorectal cancers. PloS One 2008 ; 3 : e2583. [CrossRef] [PubMed] [Google Scholar]
- Kim TM, Laird PW, Park PJ. The landscape of microsatellite instability in colorectal and endometrial cancer genomes. Cell 2013 ; 155 : 858–868. [CrossRef] [PubMed] [Google Scholar]
- Jonchere V, Marisa L, Greene M, et al. Identification of positively and negatively selected driver gene mutations associated with colorectal cancer with microsatellite instability. Cell Mol Gastroenterol Hepatol 2018 ; 6 : 277–300. [Google Scholar]
- Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer; 5 : 761–72. [Google Scholar]
- Yoshino I, Goedegebuure PS, Peoples GE, et al. Human tumor-infiltrating CD4+ T cells react to B cell lines expressing heat shock protein 70. J Immunol 1994 ; 153 : 4149–4158. [PubMed] [Google Scholar]
- Dorard C, de Thonel A, Collura A, et al. Expression of a mutant HSP110 sensitizes colorectal cancer cells to chemotherapy and improves disease prognosis. Nat Med 2011 ; 17 : 1283–1289. [CrossRef] [PubMed] [Google Scholar]
- Collura A, Lagrange A, Svrcek M, et al. Patients with colorectal tumors with microsatellite instability and large deletions in HSP110 T17 have improved response to 5-fluorouracil-based chemotherapy. Gastroenterology 2014; 146 : 401–11e1. [CrossRef] [PubMed] [Google Scholar]
- Berthenet K, Bokhari A, Lagrange A, et al. HSP110 promotes colorectal cancer growth through STAT3 activation. Oncogene 2017 ; 36 : 2328–2336. [Google Scholar]
- Sargent DJ, Marsoni S, Monges G, et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J Clin Oncol 2010 ; 28 : 3219–3226. [CrossRef] [PubMed] [Google Scholar]
- Gryfe R, Kim H, Hsieh ET, et al. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N Engl J Med 2000 ; 342 : 69–77. [Google Scholar]
- Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol 2005 ; 23 : 609–618. [CrossRef] [PubMed] [Google Scholar]
- Andre T, Boni C, Mounedji-Boudiaf L, et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N Engl J Med 2004 ; 350 : 2343–2351. [Google Scholar]
- Elsaleh H, Joseph D, Grieu F, et al. Association of tumour site and sex with survival benefit from adjuvant chemotherapy in colorectal cancer. Lancet 2000 ; 355 : 1745–1750. [CrossRef] [PubMed] [Google Scholar]
- Zaanan A, Costes L, Gauthier M, et al. Chemotherapy of advanced small-bowel adenocarcinoma: a multicenter AGEO study. Ann Oncol 2010 ; 21 : 1786–1793. [CrossRef] [PubMed] [Google Scholar]
- Tougeron D, Mouillet G, Trouilloud I, et al. Efficacy of adjuvant chemotherapy in colon cancer with microsatellite instability: A large multicenter AGEO study. J Natl Cancer Inst 2016; 108. doi: 10.1093/jnci/djv438. [Google Scholar]
- Tougeron D, Sickersen G, Mouillet G, et al. Predictors of disease-free survival in colorectal cancer with microsatellite instability: An AGEO multicentre study. Eur J Cancer 2015 ; 51 : 925–934. [CrossRef] [PubMed] [Google Scholar]
- Venderbosch S, Nagtegaal ID, Maughan TS, et al. Mismatch repair status and BRAF mutation status in metastatic colorectal cancer patients: a pooled analysis of the CAIRO, CAIRO2, COIN, and FOCUS studies. Clin Cancer Res 2014 ; 20 : 5322–5330. [CrossRef] [PubMed] [Google Scholar]
- Overman MJ, Lonardi S, Wong KYM, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J Clin Oncol 2018 ; 36 : 773–779. [CrossRef] [PubMed] [Google Scholar]
- Cohen R, Hain E, Buhard O, et al. Association of primary resistance to immune checkpoint inhibitors in metastatic colorectal cancer with misdiagnosis of microsatellite instability or mismatch repair deficiency status. JAMA Oncol 2018; Nov 15. doi: 10.1001/jamaoncol.2018.4942. [Google Scholar]
- Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer 2012 ; 12 : 298–306. [Google Scholar]
- Bokhari A, Jonchere V, Lagrange A, et al. Targeting nonsense-mediated mRNA decay in colorectal cancers with microsatellite instability. Oncogenesis 2018 ; 7 : 70. [CrossRef] [PubMed] [Google Scholar]
- Maby P, Tougeron D, Hamieh M, et al. Correlation between density of CD8+ T-cell infiltrate in microsatellite unstable colorectal cancers and frameshift mutations: a rationale for personalized immunotherapy. Cancer Res 2015 ; 75 : 3446–3455. [Google Scholar]
- Mlecnik B, Bindea G, Angell HK, et al. Integrative analyses of colorectal cancer show immunoscore is a stronger predictor of patient survival than microsatellite instability. Immunity 2016 ; 44 : 698–711. [CrossRef] [PubMed] [Google Scholar]
- Marisa L, Svrcek M, Collura A, et al. The balance between cytotoxic T-cell lymphocytes and immune checkpoint expression in the prognosis of colon tumors. J Natl Cancer Inst 2018 ; 01 1 : 110.10.1093/jnci/djx136 [Google Scholar]
Liste des tableaux
Caractéristiques pronostiques et de réponse à la chimiothérapie des CCR (MSI versus MSS). MSI : instabilité des microsatellites ; MSS : stabilité des microsatellites ; CT : chimiothérapie ; anti-ICK : anti-immunocheckpoint.
Liste des figures
|
Figure 1. Étapes de la tumorigenèse MSI. MSI : instabilité des microsatellites ; ICK : intestinal cell kinase. |
| Dans le texte | |
|
Figure 2. Algorithme pour la décision thérapeutique de chimiothérapie adjuvante dans les CCR de stade 2-3. CT : chimiothérapie. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.