")
")
| Issue |
Med Sci (Paris)
Volume 34, Number 5, Mai 2018
|
|
|---|---|---|
| Page(s) | 417 - 423 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/20183405014 | |
| Published online | 13 June 2018 | |
L’adiponectine
Un anti-inflammatoire et anti-dépresseur endogène ?
Adiponectin: an endogenous molecule with anti-inflammatory and antidepressant properties?
Université Côte d’Azur, CNRS, Institut de Pharmacologie Moléculaire et Cellulaire, UMR7275, 660, route des Lucioles, Sophia Antipolis, 06560 Valbonne, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
L’adiponectine (ApN) est une hormone produite par le tissu adipeux dont le taux plasmatique est diminué chez les personnes en surpoids ou obèses ainsi que chez les patients diabétiques. En périphérie, cette baisse du taux circulant d’ApN induit l’établissement d’un état inflammatoire chronique à bas bruit, le développement d’une résistance à l’insuline et de plaques d’athérome. Inversement, des conditions de vie « favorables », la perte de poids et la pratique régulière d’exercice physique permettent d’augmenter la concentration sanguine d’ApN. Certaines formes d’ApN peuvent gagner le cerveau par le biais du liquide cérébrospinal. À ce niveau, l’augmentation de l’ApN exerce de puissants effets anti-dépresseurs et anxiolytiques, notamment en réduisant la neuroinflammation.
Abstract
Adiponectin (ApN) is a hormone produced by adipose tissue, yet the plasma level of ApN is decreased in overweight and obese people, as well as in people with diabetes. In the periphery, this decrease in circulating levels of ApN induces the establishment of a chronic low-grade inflammatory state and is involved in the development of insulin resistance and atheromas. Conversely, “favorable” living conditions, weight loss and regular physical exercise increase ApN blood concentration. Some forms of ApN can reach the brain parenchyma through the cerebrospinal fluid. In the brain, the increase in ApN exerts powerful antidepressant and anxiolytic effects, in particular by fighting against neuroinflammation.
© 2018 médecine/sciences – Inserm
L’adiponectine : une hormone du tissu adipeux présente dans le cerveau
Dans les années 1990, l’adiponectine (ApN) a été identifiée presque simultanément par quatre groupes de recherche qui ont baptisé la protéine de différents noms : Acrp30 (adipocyte complement-related protein of 30 kDa) [1], apM1 (adipose most abundant gene transcript 1) [2], AdipoQ [3] et GBP28 (gelatin-binding protein of 28 kDa) [4]. Désormais le terme d’adiponectine est le plus utilisé pour désigner cette protéine.
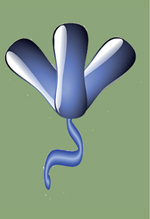
L’ApN, une hormone sécrétée essentiellement par les adipocytes, est une protéine très abondante dans le plasma (entre 5 et 30 µg/ml). Il s’agit d’une adipokine, c’est-à-dire une cytokine (hormone jouant un rôle dans le système immunitaire) produite par le tissu adipeux. Contrairement à la leptine, une autre adipokine libérée par les adipocytes lorsqu’ils sont gonflés de gras, et qui joue le rôle d’une hormone de satiété, l’ApN est libérée par les adipocytes lorsque leurs stocks de gras sont utilisés. L’ApN est une protéine de 30 kDa comprenant un domaine globulaire à son extrémité C-terminale, et un domaine de type collagène. Le domaine globulaire possède la même séquence en acides aminés que le facteur du complément C1q et de fortes similitudes avec les domaines globulaires d’autres protéines telles que, par exemple, les collagènes VIII et X, et le TNF-α (tumor necrosis factor a) [5]. Le domaine collagène permet la formation de trimères, d’hexamères et d’autres formes multimériques sécrétées d’ApN. La forme monomérique n’est pas libérée dans la circulation. Elle n’est détectée que dans les adipocytes. La forme globulaire circulante, appelée gApN, est produite par clivage protéolytique par l’élastase leucocytaire de la forme entière monomérique [6]. Les formes trimériques et hexamériques sont respectivement considérées comme les formes de faible et moyenne masses moléculaires. Les gros complexes (formés de 18 unités et plus) sont de haute masse moléculaire. Dans la circulation, les formes hexamériques et de haute masse moléculaire sont majoritaires (Figure 1).
 |
Figure 1. Représentation schématique de la structure de l’adiponectine et de ses oligomères. La forme entiere de l’adiponectine comprend 244 acides amines, un domaine de type collagene vers l’extremite N-terminale et un domaine globulaire de type C1q a son extremite C-terminale. Dans le plasma, les monomeres d’adiponectine se combinent par leur domaine globulaire pour former des complexes multimeriques tels que des trimeres, des hexameres ou des gros multimeres de haute masse moleculaire. Il existe egalement une forme globulaire circulante de l’adiponectine. |
La présence d’ApN dans le cerveau a longtemps été controversée. Toutefois, il est désormais admis que certaines formes d’ApN sont capables de franchir la barrière hémato-encéphalique (BHE). Une étude a notamment montré qu’une injection par voie intraveineuse ou intracérébroventriculaire (icv) d’ApN agit au niveau central et réduit la glycémie, la lipidémie et le poids corporel des souris [7]. En 2007, Kubota et al. ont confirmé l’action de l’ApN dans le cerveau, en montrant que l’injection périphérique d’ApN augmentait la prise alimentaire et diminuait les dépenses énergétiques des souris [8]. Depuis, plusieurs études ont détecté l’ApN dans le liquide cérébrospinal (LCS) murin et humain, à des taux 100 fois plus faibles que les taux plasmatiques [9–11]. Dans le cerveau, contrairement à la circulation, les formes actives seraient celles de faible masse moléculaire, plus particulièrement les formes trimériques qui seraient les seules à pouvoir franchir la BHE. Enfin, la présence d’ARN messagers (ARNm) de l’ApN a été détectée dans des extraits de cerveau de souris et de poulets, suggérant qu’il pourrait exister une production locale de l’hormone [12, 13]. Chez l’homme, les ARNm de l’ApN ont été détectés uniquement dans l’hypophyse [14, 15].
L’ApN agit par l’intermédiaire de ses deux principaux récepteurs : l’AdipoR1 et l’AdipoR2, qui possèdent 67 % d’homologie [16]. Ces récepteurs contiennent 7 domaines transmembranaires, comme les récepteurs couplés aux protéines G, avec une topologie membranaire inversée, l’extrémité N-terminale étant intracellulaire et l’extrémité C-terminale, extracellulaire [16]. Les récepteurs AdipoR1 et R2 ont des expressions tissulaires distinctes ainsi que des affinités de liaison différentes pour les formes globulaire et entière de l’ApN. Les deux récepteurs sont exprimés dans le cerveau, en particulier dans l’hypothalamus et le gyrus denté de l’hippocampe [8, 9, 17]. Leur expression a été rapportée au niveau neuronal [8, 9], astrocytaire [18] et microglial [19].
Obésité, troubles psychiques et adiponectine
De nombreux arguments ont permis d’établir un lien entre les maladies métaboliques à composante inflammatoire (comme le surpoids et l’obésité) et la dépression. L’obésité et la dépression sont deux pathologies distinctes du point de vue étio-pathologique, mais leurs prévalences croissent parallèlement dans le monde. Des relations bidirectionnelles existent entre ces deux maladies : la dépression augmente le risque de prise de poids et d’obésité, et inversement. Par exemple, l’incidence de la dépression chez les individus obèses est proche de 30 %, ce qui est significativement plus élevé que dans la population générale (où elle se situe autour de 7,5 %). Ces observations suggèrent l’existence de mécanismes physiopathologiques communs parmi lesquels l’inflammation tiendrait une position centrale [20]. Notons que les symptômes dépressifs que développent les personnes obèses présentent souvent des caractéristiques atypiques (augmentation de l’appétit, prise de poids, hypersomnie, fatigue, etc.) [21], suggérant qu’il existerait un sous-type particulier de dépression qui serait lié à l’état métabolique et inflammatoire.
Quels sont les liens entre l’ApN et la dépression ? Chez l’homme, la corrélation entre les taux plasmatiques d’ApN et la dépression est controversée, probablement à cause de l’existence de différents sous-types de dépression. En effet, certains rapports indiquent que les taux sériques d’ApN sont plus élevés chez les sujets souffrant de dépression majeure par rapport à des témoins sains [22]. D’autres études montrent des niveaux inférieurs [23] ou inchangés [24]. Récemment, il a été démontré que des taux faibles d’ApN étaient associés à une évolution défavorable des troubles bipolaires [25].
Chez les rongeurs, les résultats sont plus unanimes. En 2012, Liu et al. ont montré que les niveaux d’ApN dans le plasma sont réduits dans un modèle murin de dépression induit par un stress chronique (modèle de stress social). Chez les souris hétérozygotes ApN+/- , la réduction des taux d’ApN exacerbe les comportements dépressifs, qui se manifestent par une augmentation de l’aversion sociale, de l’anhédonie et de la résignation. De plus, la neutralisation des effets centraux de l’ApN, par injection icv d’anticorps spécifiques, provoque un comportement dépressif chez les animaux. Inversement, l’injection icv d’ApN exogène produit des effets antidépresseurs chez les souris saines ou diabétiques et obèses. Ces résultats suggèrent donc un rôle important de l’ApN dans la régulation des comportements dépressifs chez la souris [9].
Récemment, l’implication de l’ApN, via ses récepteurs AdipoR2, a été révélée dans les phénomènes d’extinction de la mémoire de peur associée à un contexte. Ces résultats suggèrent que l’ApN pourrait moduler les processus impliqués dans le stress post-traumatique [26]. Une étude récente confirme d’ailleurs qu’une baisse des taux circulants d’ApN est associée à la dépression liée à ce type de stress [27].
En 2014, une autre étude a mis en évidence un rôle de l’ApN dans la dépression. Yau et al. ont en effet montré que les effets antidépresseurs de l’exercice physique reposaient sur l’ApN [10]. L’ApN participe également aux effets antidépresseurs de l’environnement enrichi (EE), un modèle expérimental qui stimule les activités sociales, motrices, sensitives et cognitives des souris [28]. En effet, les souris déficientes en ApN sont plus sensibles aux effets dépresseurs d’un traitement chronique à la corticostérone que des souris sauvages, et sont partiellement insensibles aux effets antidépresseurs de l’EE. Chez les souris sauvages, l’EE induit une augmentation sélective de la concentration des formes de basse et faible masses moléculaires d’ApN dans le LCS des souris rendues « dépressives ». De plus, l’injection par voie intraveineuse de gApN exogène exerce des effets antidépresseurs rapides et efficaces sur les souris ; l’un des mécanismes d’action possibles serait une activation des neurones hypothalamiques [28].
L’inflammation comme lien entre ApN et dépression
ApN et inflammation périphérique
En périphérie, un faible taux circulant d’ApN est associé à différentes maladies inflammatoires chroniques comme l’obésité, le diabète de type 2 et la résistance à l’insuline, de même que l’athérosclérose, l’hypertension et les maladies coronariennes [3, 29]. À l’inverse, un niveau d’ApN sérique élevé a été observé dans d’autres pathologies telles que le lupus érythémateux disséminé [30], la mucoviscidose [31], les maladies inflammatoires de l’intestin [32] et la polyarthrite rhumatoïde [33]. Le risque d’infarctus du myocarde est diminué d’un facteur deux, et le risque de calcification des artères coronaires d’un facteur sept, chez les sujets dont le taux sérique d’ApN est le plus élevé par rapport à des sujets présentant des taux moyens [34]. Une hypothèse permettant de concilier ces résultats divergents serait que l’augmentation de l’ApN que l’on observe pourrait être, dans certains cas, une réaction de l’organisme visant à contrebalancer l’inflammation chronique caractéristique de ces maladies.
Plusieurs mécanismes ont été suggérés pour expliquer l’implication de l’ApN dans les processus inflammatoires, dont des actions directes sur les cellules médiatrices de l’inflammation, sur l’activation de la voie NF-κB (nuclear factor-kappa B), et l’interaction avec le TNF-α.
Les cellules mononucléées sanguines (monocytes, lymphocytes T et B, et NK [natural killer]) expriment à leur surface les récepteurs de l’ApN, AdipoR1 et R2 [35]. Dans un contexte d’athérosclérose, caractérisée par une inflammation chronique, l’ApN inhibe la croissance des progéniteurs myélomonocytaires et le fonctionnement des macrophages matures [36]. Elle inhibe, en particulier, la transformation des macrophages en cellules spumeuses (qui participent au développement de l’athérosclérose) [37], et stimule la production d’IL(interleukine)-10, une cytokine anti-inflammatoire [38]. En réduisant l’expression de molécules d’adhérence telles que VCAM-1 (vascular cell adhesion molecule-1), ICAM-1 (intercellular adhesion molecule-1), de l’E-sélectine et de l’IL-8 par les cellules endothéliales aortiques humaines, l’ApN empêcherait également l’adhérence des monocytes à l’endothélium [39]. Dans les macrophages, l’ApN inhibe l’activation du facteur NF-κB induite par une interaction avec les TLR (Toll like receptors) [40] et dans les neutrophiles, elle réduit la production des ROS (reactive oxygen species) [41].
Le facteur nucléaire NF-κB est un acteur clé dans la réponse inflammatoire. L’action de l’ApN sur la voie de signalisation du NF-κB est complexe : elle peut en effet être à la fois activatrice et inhibitrice sur les cellules myéloïdes. Dans les macrophages, l’ApN inhibe la voie NF-κB induite par le TNF-α [42]. Elle serait donc anti-inflammatoire vis-à-vis des réponses impliquant le TNF-α. La relation ApN/TNF-α serait en fait bi-directionnelle. En effet, in vitro, dans des cultures d’adipocytes humains et murins, le TNF-α inhibe l’expression et la sécrétion de l’ApN [43]. Inversement, in vivo, des taux élevés d’ARNm du TNF-α sont retrouvés dans le tissu adipeux et le plasma de souris déficientes en ApN. L’expression d’ApN, par l’utilisation d’un vecteur viral, rétablit chez ces souris déficientes des niveaux physiologiques de TNF-α [29]. Dans les macrophages, l’ApN inhibe fortement l’expression des ARNm du TNF-α induite par le lipopolysaccharide (LPS), un composant des parois bactériennes à Gram négatif couramment utilisé pour provoquer une réponse inflammatoire dépendant du TLR4 [36]. Chez l’homme, les taux plasmatiques de TNF-α et d’ApN mesurés chez des patients dépressifs en rémission, et ayant reçu un traitement antidépresseur, sont respectivement plus faibles (pour le TNF-α) et plus élevés (pour l’ApN) que chez les patients non traités [44]. Anderson et al. ont exploré la relation entre inflammation aiguë et taux d’adipokines au sein d’une cohorte humaine de 20 patients en bonne santé. L’endotoxémie, c’est-à-dire la présence d’endotoxines, dont le LPS, dans le sang, a été associée à une augmentation de la concentration de TNF-α et d’IL-6 dans l’ensemble des leucocytes du sang, ainsi que dans le tissu gras, mais sans modification significative des taux plasmatiques d’ApN. En revanche, une diminution d’expression des récepteurs AdipoR1 et R2 a été observée, suggérant que les fonctions de sensibilisation à l’insuline et de régulation de l’inflammation par l’ApN étaient altérées [45]. Dans des modèles animaux de choc septique par injection de LPS, la déficience en ApN est associée à une augmentation marquée du statut inflammatoire et à une exacerbation des lésions hépatiques [46]. Chez l’homme, une corrélation inverse similaire a été observée dans une étude, menée en 2010, sur 33 patients atteints de sepsis sévère ou de choc septique induit par une infection bactérienne systémique [47]. La modulation de l’ApN dans le sepsis pourrait donc représenter une cible thérapeutique potentielle pour atténuer la réponse inflammatoire périphérique en cas d’infection bactérienne sévère.
Neuroinflammation, dépression et adiponectine
Il y a une dizaine d’années, l’hypothèse de l’implication de la neuroinflammation dans la dépression a été évoquée. Cette théorie s’est fondée sur une étude révélant que l’administration de cytokines pro-inflammatoires à des volontaires sains provoquait des symptômes de type dépressif [48]. La production excessive de médiateurs de l’inflammation par les macrophages jouerait, selon cette hypothèse, un rôle essentiel dans la pathogenèse de la dépression. D’autres travaux, montrant une activation du système immunitaire inné chez certains patients dépressifs, ont confirmé ces observations [49].
Malgré une littérature abondante permettant de relier ApN et inflammation périphérique, peu d’études se sont intéressées aux effets de l’hormone sur la neuroinflammation. En 2013, Piccio et al. ont montré que des souris déficientes en ApN étaient plus sensibles à l’induction d’une sclérose en plaque (SEP) par injection d’antigènes de la myéline, dans le modèle de l’encéphalomyélite allergique expérimentale (EAE), avec une réponse inflammatoire accrue dans la moelle épinière [50]. À l’inverse, un traitement d’une semaine par injections intrapéritonéales quotidiennes de gApN ralentit la progression de la maladie et réduit les niveaux de cytokines pro-inflammatoires dans la moelle épinière.
Dans le SNC, la réponse inflammatoire se caractérise par la présence, dans le parenchyme du cerveau, de cellules microgliales activées et d’astrocytes réactifs, mais aussi par une implication directe du système immunitaire adaptatif, une production accrue de cytokines, de chimiokines et de prostaglandines, l’activation de la cascade des protéines du complément, et la production d’espèces réactives de l’oxygène et de l’azote (ROS/RNS), qui, dans certains cas, peuvent conduire à la rupture de la BHE.
Au niveau central, l’ApN exercerait de puissants effets anti-inflammatoires en modulant le phénotype de la microglie. Les cellules microgliales maintiennent l’homéostasie neuronale et sont impliquées dans la veille immunitaire en sondant en permanence l’environnement cérébral grâce à des prolongements cellulaires. Exprimant de nombreux canaux ioniques, des récepteurs TLR et des récepteurs de chimiokines, et produisant des cytokines, ces cellules réagissent rapidement en réponse à des altérations subtiles de leur microenvironnement comme des modifications d’homéostasie ionique et des dommages cérébraux induits par des agrégats protéiques ou des agents pathogènes. Comme les macrophages, la microglie peut schématiquement adopter deux types de phénotype, M1 et M2. Le phénotype M1 de la microglie est lié à l’induction d’une réponse inflammatoire, alors que le phénotype M2 est associé à une activité neuroprotectrice. La polarisation de la microglie est importante dans l’homéostasie du SNC. Elle est en effet impliquée dans les processus d’apprentissage et de mémoire [51].
L’absence d’ApN exacerbe les réponses neuroinflammatoires. Des souris déficientes en ApN sont en effet plus sensibles à la neuroinflammation induite par une injection périphérique de LPS. Une augmentation des niveaux de cytokines proinflammatoires plus importante est observée dans différentes zones du cerveau et dans le LCS de ces souris par rapport à des souris exprimant l’ApN. L’absence d’ApN se traduit également par une plus forte infiltration de monocytes dans le cerveau des souris traitées au LPS. Cette réponse exacerbée des souris déficientes en ApN à un stimulus pro-inflammatoire résulte d’une réactivité accrue de leur microglie. La microglie des souris déficientes en ApN exprime en effet plus d’ARNm et de protéines pro-inflammatoires, après traitement au LPS, que celle issue de cerveaux de souris sauvages [19].
Si, in vivo, des effets combinés sur différentes populations cellulaires et/ou des effets indirects ne peuvent être exclus, nous avons montré que l’ApN, sous ses formes globulaire et entière, exerçait des effets anti-inflammatoires directs, préventifs et curatifs sur la microglie. Le traitement par la gApN permet en effet de réduire la sécrétion de cytokines pro-inflammatoires par la microglie préalablement activée in vivo. La gApN limite également la sécrétion de cytokines pro-inflammatoires par la microglie traitée ex vivo par du LPS [19]. Ces effets anti-inflammatoires de l’ApN sont spécifiques de la microglie : la gApN n’a pas d’effet sur des astrocytes murins stimulés par du LPS en culture primaire [19]. Sur la lignée murine de cellules microgliales BV2, la gApN réduit, de manière dose-dépendante, l’expression des ARNm et des protéines pro-inflammatoires et les stress oxydatif et nitrosatif engendrés par le traitement au LPS. L’ApN agit sur la microglie via son récepteur AdipoR1 et induit une voie de signalisation impliquant l’AMPK (AMP-activated protein kinase). Enfin, elle interférerait également avec la translocation de la sous-unité p65 de NF-κB induite par le LPS, ce qui expliquerait ses effets anti-inflammatoires (Figure 2) [19].
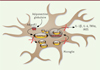 |
Figure 2. Représentation schématique des mécanismes d’action de l’adiponectine globulaire sur la microglie. L’adiponectine globulaire, en se liant a son recepteur AdipoR1 present a la surface des cellules microgliales, stimule l’activation de l’AMPKα (AMP-activated protein kinase) en favorisant la phosphorylation du residu threonine en position 172. Ceci empeche la degradation de la kinase I-B et la translocation de la sous-unite p65 de NF-B (nuclear factor-kappa B) dans le noyau, ou elle se lie a l’ADN sur des sequences consensus presentes dans les promoteurs de nombreux genes codant des proteines pro-inflammatoires comme les cytokines IL(interleukine)-1, IL-6 et TNF- tumor necrosisfactor-). L’adiponectine limite egalement l’expression de la iNOS (inducible nitric oxide synthase) et la liberation de monoxyde d’azote. |
L’adiponectine, un médiateur des effets anti-dépresseurs de l’environnement enrichi
Tous les individus ne sont pas égaux vis-à-vis de leur susceptibilité à développer des troubles psychiatriques. Par exemple, des abus et violences au cours de l’enfance, ou des conditions de vie socio-économiquement difficiles, favorisent l’apparition de désordres mentaux à l’âge adulte [52]. De même, des susceptibilités génétiques prédisposant aux épisodes dépressifs, comme des polymorphismes dans les gènes codant les récepteurs de la sérotonine [53] ou du BDNF (brain-derived neurotrophic factor) [54], ont été rapportées.
Chez la souris, des conditions de vie favorables, comme celles procurées par l’environnement enrichi, se traduisent par une augmentation dans le LCS des taux des formes de basse masse moléculaire de l’ApN. Son passage au travers de la BHE serait facilité dans ces conditions. Le mécanisme impliqué reste cependant encore inconnu. Dans un modèle de souris rendues dépressives, l’environnement enrichi permet d’atténuer l’augmentation des taux d’ARNm de cytokines pro-inflammatoires (IL-1β, IL-6 et TNF-α) au niveau de l’hypothalamus et de l’hippocampe, deux zones du cerveau étroitement liées à la dépression. Une orientation du profil d’activation de la microglie vers un profil anti-inflammatoire de type M2 est stimulée chez des souris en environnement enrichi, comparées à des souris hébergées en conditions standard, qui, elles, présentent un phénotype pro-inflammatoire. Les effets anti-inflammatoires sur la microglie et les effets comportementaux antidépresseurs de l’environnement enrichi reposent, au moins en partie, sur l’ApN. L’injection icv de gApN mime en effet les effets anti-inflammatoires de l’environnement enrichi sur le profil d’activation de la microglie avec l’induction d’une diminution de l’expression et de la production des cytokines pro-inflammatoires, tout en augmentant les niveaux des ARNm de marqueurs anti-inflammatoires comme l’Arg1 (Arginase 1) et l’IL-10, et améliore les comportements anxio-dépressifs des souris [11].
Conclusion
L’ApN est une adipokine produite par le tissu gras, aux propriétés antidépressives, dont l’action dans le cerveau serait favorisée par des conditions de vie positives et stimulantes, combinant activités physiques, cognitives et sociales. Les propriétés antidépressives de l’ApN peuvent être attribuées, notamment, à ses effets anti-inflammatoires directs sur la microglie, et donc à sa capacité à limiter la neuroinflammation. Le développement d’agonistes sélectifs des récepteurs de l’ApN pourrait ainsi représenter une stratégie thérapeutique alternative prometteuse pour réduire la neuroinflammation dans de nombreuses maladies cérébrales comme les maladies neurodégénératives et les troubles dépressifs (Figure 3).
 |
Figure 3. Schéma illustrant comment l’environnement enrichi pourrait lutter contre les comportements anxio-dépressifs induits par le stress. La liberation chronique dans le sang de corticosterone, hormone de stress produite par les glandes surrenales, agit au niveau cerebral en promouvant une activation pro-inflammatoire de la microglie, induisant ainsi l’etablissement d’un etat neuroinflammatoire et l’apparition de comportements anxio-depressifs. A l’inverse, des conditions de vie favorables ou se melent activites physiques, sociales et cognitives, telles que celles fournies par l’environnement enrichi, permettent l’augmentation des taux d’adiponectine, adipokine produite par le tissu adipeux, qui va promouvoir dans le cerveau un phenotype microglial antiinflammatoire et ainsi lutter contre la neuroinflammation et les comportements anxio-depressifs. |
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Références
- Scherer PE, Williams S, Fogliano M, et al. A novel serum protein similar to C1q, produced exclusively in adipocytes. J Biol Chem 1995 ; 270 : 26746–26749. [CrossRef] [PubMed] [Google Scholar]
- Maeda K, Okubo K, Shimomura I, et al. cDNA cloning and expression of a novel adipose specific collagen-like factor, apM1 (adiPose most abundant gene transcript 1). Biochem Biophys Res Commun 1996 ; 221 : 286–289. [Google Scholar]
- Hu E, Liang P, Spiegelman BM. AdipoQ is a novel adipose-specific gene dysregulated in obesity. J Biol Chem 1996 ; 271 : 10697–10703. [CrossRef] [PubMed] [Google Scholar]
- Nakano Y, Tobe T, Choi-Miura NH, et al. Isolation and characterization of GBP28, a novel gelatin-binding protein purified from human plasma. J Biochem 1996 ; 120 : 803–812. [CrossRef] [PubMed] [Google Scholar]
- Berg AH, Combs TP, Scherer PE. ACRP30/adiponectin: an adipokine regulating glucose and lipid metabolism. Trends Endocrinol Metab 2002 ; 13 : 84–89. [CrossRef] [PubMed] [Google Scholar]
- Waki H, Yamauchi T, Kamon J, et al. Generation of globular fragment of adiponectin by leukocyte elastase secreted by monocytic cell line THP-1. Endocrinology 2005 ; 146 : 790–796. [CrossRef] [PubMed] [Google Scholar]
- Qi Y, Takahashi N, Hileman SM, et al. Adiponectin acts in the brain to decrease body weight. Nat Med 2004 ; 10 : 524–529. [CrossRef] [PubMed] [Google Scholar]
- Kubota N, Yano W, Kubota T, et al. Adiponectin stimulates AMP-activated protein kinase in the hypothalamus and increases food intake. Cell Metab 2007 ; 6 : 55–68. [CrossRef] [PubMed] [Google Scholar]
- Liu J, Guo M, Zhang D, et al. Adiponectin is critical in determining susceptibility to depressive behaviors and has antidepressant-like activity. Proc Natl Acad Sci USA 2012 ; 109 : 12248–12253. [CrossRef] [Google Scholar]
- Yau SY, Li A, Hoo RL, et al. Physical exercise-induced hippocampal neurogenesis and antidepressant effects are mediated by the adipocyte hormone adiponectin. Proc Natl Acad Sci USA 2014 ; 111 : 15810–15815. [CrossRef] [Google Scholar]
- Chabry J, Nicolas S, Cazareth J, et al. Enriched environment decreases microglia and brain macrophages inflammatory phenotypes through adiponectin-dependent mechanisms: relevance to depressive-like behavior. Brain Behav Immun 2015 ; 50 : 275–287. [CrossRef] [PubMed] [Google Scholar]
- Maddineni S, Metzger S, Ocon O, et al. Adiponectin gene is expressed in multiple tissues in the chicken: food deprivation influences adiponectin messenger ribonucleic acid expression. Endocrinology 2005 ; 146 : 4250–4256. [CrossRef] [PubMed] [Google Scholar]
- Hoyda TD, Fry M, Ahima RS, Ferguson AV. Adiponectin selectively inhibits oxytocin neurons of the paraventricular nucleus of the hypothalamus. J Physiol 2007 ; 585 : 805–816. [CrossRef] [PubMed] [Google Scholar]
- Rodriguez-Pacheco F, Martinez-Fuentes AJ, Tovar S, et al. Regulation of pituitary cell function by adiponectin. Endocrinology 2007 ; 148 : 401–410. [CrossRef] [PubMed] [Google Scholar]
- Wilkinson M, Brown R, Imran SA, Ur E. Adipokine gene expression in brain and pituitary gland. Neuroendocrinology 2007 ; 86 : 191–209. [Google Scholar]
- Yamauchi T, Kamon J, Ito Y, et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 2003 ; 423 : 762–769. [CrossRef] [PubMed] [Google Scholar]
- Suyama S, Lei W, Kubota N, et al. Adiponectin at physiological level glucose-independently enhances inhibitory postsynaptic current onto NPY neurons in the hypothalamic arcuate nucleus. Neuropeptides 2017 ; 65 : 1–9. [CrossRef] [PubMed] [Google Scholar]
- Wan Z, Mah D, Simtchouk S, et al. Globular adiponectin induces a pro-inflammatory response in human astrocytic cells. Biochem Biophys Res Commun 2014 ; 446 : 37–42. [Google Scholar]
- Nicolas S, Cazareth J, Zarif H, et al. Globular adiponectin limits microglia pro-inflammatory phenotype through an AdipoR1/NF-kappaB signaling pathway. Front Cell Neurosci 2017 ; 11 : 352. [PubMed] [Google Scholar]
- Capuron L, Lasselin J, Castanon N. Role of adiposity-driven inflammation in depressive morbidity. Neuropsychopharmacology 2017 ; 42 : 115–128. [CrossRef] [PubMed] [Google Scholar]
- Lojko D, Buzuk G, Owecki M, et al. Atypical features in depression: association with obesity and bipolar disorder. J Affect Disord 2015 ; 185 : 76–80. [CrossRef] [PubMed] [Google Scholar]
- Jow GM, Yang TT, Chen CL. Leptin and cholesterol levels are low in major depressive disorder, but high in schizophrenia. J Affect Disord 2006 ; 90 : 21–27. [CrossRef] [PubMed] [Google Scholar]
- Cizza G, Nguyen VT, Eskandari F, et al. Low 24-hour adiponectin and high nocturnal leptin concentrations in a case-control study of community-dwelling premenopausal women with major depressive disorder: the premenopausal, osteopenia/osteoporosis, women, alendronate, depression (Power) study. J Clin Psychiatry 2010 ; 71 : 1079–1087. [CrossRef] [PubMed] [Google Scholar]
- Carvalho AF, Rocha DQ, McIntyre RS, et al. Adipokines as emerging depression biomarkers: a systematic review and meta-analysis. J Psychiatr Res 2014 ; 59 : 28–37. [Google Scholar]
- Mansur RB, Rizzo LB, Santos CM, et al. Adipokines, metabolic dysfunction and illness course in bipolar disorder. J Psychiatr Res 2016 ; 74 : 63–69. [Google Scholar]
- Zhang D, Wang X, Wang B, et al. Adiponectin regulates contextual fear extinction and intrinsic excitability of dentate gyrus granule neurons through AdipoR2 receptors. Mol Psychiatry 2017 ; 22 : 1044–1055. [CrossRef] [PubMed] [Google Scholar]
- Na KS, Kim EK, Park JT. Decreased plasma adiponectin among male firefighters with symptoms of post-traumatic stress disorder. J Affect Disord 2017 ; 221 : 254–258. [CrossRef] [PubMed] [Google Scholar]
- Nicolas S, Veyssiere J, Gandin C, et al. Neurogenesis-independent antidepressant-like effects of enriched environment is dependent on adiponectin. Psychoneuroendocrinology 2015 ; 57 : 72–83. [CrossRef] [PubMed] [Google Scholar]
- Maeda N, Shimomura I, Kishida K, et al. Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat Med 2002 ; 8 : 731–737. [CrossRef] [PubMed] [Google Scholar]
- Rovin BH, Song H, Hebert LA, et al. Plasma, urine, and renal expression of adiponectin in human systemic lupus erythematosus. Kidney Int 2005 ; 68 : 1825–1833. [CrossRef] [PubMed] [Google Scholar]
- Moriconi N, Kraenzlin M, Muller B, et al. Body composition and adiponectin serum concentrations in adult patients with cystic fibrosis. J Clin Endocrinol Metab 2006 ; 91 : 1586–1590. [CrossRef] [PubMed] [Google Scholar]
- Karmiris K, Koutroubakis IE, Xidakis C, et al. Circulating levels of leptin, adiponectin, resistin, and ghrelin in inflammatory bowel disease. Inflamm Bowel Dis 2006 ; 12 : 100–105. [CrossRef] [PubMed] [Google Scholar]
- Otero M, Lago R, Gomez R, et al. Changes in plasma levels of fat-derived hormones adiponectin, leptin, resistin and visfatin in patients with rheumatoid arthritis. Ann Rheum Dis 2006 ; 65 : 1198–1201. [CrossRef] [PubMed] [Google Scholar]
- Pischon T, Girman CJ, Hotamisligil GS, et al. Plasma adiponectin levels and risk of myocardial infarction in men. JAMA 2004 ; 291 : 1730–1737. [CrossRef] [PubMed] [Google Scholar]
- Pang TT, Narendran P. The distribution of adiponectin receptors on human peripheral blood mononuclear cells. Ann NY Acad Sci 2008 ; 1150 : 143–145. [CrossRef] [Google Scholar]
- Yokota T, Oritani K, Takahashi I, et al. Adiponectin, a new member of the family of soluble defense collagens, negatively regulates the growth of myelomonocytic progenitors and the functions of macrophages. Blood 2000 ; 96 : 1723–1732. [Google Scholar]
- Ouchi N, Kihara S, Arita Y, et al. Adipocyte-derived plasma protein, adiponectin, suppresses lipid accumulation and class A scavenger receptor expression in human monocyte-derived macrophages. Circulation 2001 ; 103 : 1057–1063. [CrossRef] [PubMed] [Google Scholar]
- Kumada M, Kihara S, Ouchi N, et al. Adiponectin specifically increased tissue inhibitor of metalloproteinase-1 through interleukin-10 expression in human macrophages. Circulation 2004 ; 109 : 2046–2049. [CrossRef] [PubMed] [Google Scholar]
- Ouedraogo R, Wu X, Xu SQ, et al. Adiponectin suppression of high-glucose-induced reactive oxygen species in vascular endothelial cells: evidence for involvement of a cAMP signaling pathway. Diabetes 2006 ; 55 : 1840–1846. [CrossRef] [PubMed] [Google Scholar]
- Yamaguchi N, Argueta JG, Masuhiro Y, et al. Adiponectin inhibits Toll-like receptor family-induced signaling. FEBS Lett 2005 ; 579 : 6821–6826. [CrossRef] [PubMed] [Google Scholar]
- Magalang UJ, Rajappan R, Hunter MG, et al. Adiponectin inhibits superoxide generation by human neutrophils. Antioxid Redox Signal 2006 ; 8 : 2179–2186. [CrossRef] [PubMed] [Google Scholar]
- Wulster-Radcliffe MC, Ajuwon KM, Wang J, et al. Adiponectin differentially regulates cytokines in porcine macrophages. Biochem Biophys Res Commun 2004 ; 316 : 924–929. [Google Scholar]
- Kappes A, Loffler G. Influences of ionomycin, dibutyryl-cycloAMP and tumour necrosis factor-alpha on intracellular amount and secretion of apM1 in differentiating primary human preadipocytes. Horm Metab Res 2000 ; 32 : 548–554. [CrossRef] [PubMed] [Google Scholar]
- Narita K, Murata T, Takahashi T, et al. Plasma levels of adiponectin and tumor necrosis factor-alpha in patients with remitted major depression receiving long-term maintenance antidepressant therapy. Prog Neuropsychopharmacol Biol Psychiatry 2006 ; 30 : 1159–1162. [CrossRef] [PubMed] [Google Scholar]
- Anderson PD, Mehta NN, Wolfe ML, et al. Innate immunity modulates adipokines in humans. J Clin Endocrinol Metab 2007 ; 92 : 2272–2279. [CrossRef] [PubMed] [Google Scholar]
- Uji Y, Yamamoto H, Maeda K, et al. Adiponectin deficiency promotes the production of inflammatory mediators while severely exacerbating hepatic injury in mice with polymicrobial sepsis. J Surg Res 2010 ; 161 : 301–311. [CrossRef] [PubMed] [Google Scholar]
- Hillenbrand A, Knippschild U, Weiss M, et al. Sepsis induced changes of adipokines and cytokines - septic patients compared to morbidly obese patients. BMC Surg 2010 ; 10 : 26. [CrossRef] [PubMed] [Google Scholar]
- Smith RS. The macrophage theory of depression. Med Hypotheses 1991 ; 35 : 298–306. [Google Scholar]
- Maes M. Major depression and activation of the inflammatory response system. Adv Exp Med Biol 1999 ; 461 : 25–46. [CrossRef] [PubMed] [Google Scholar]
- Piccio L, Cantoni C, Henderson JG, et al. Lack of adiponectin leads to increased lymphocyte activation and increased disease severity in a mouse model of multiple sclerosis. Eur J immunol 2013 ; 43 : 2089–2100. [CrossRef] [PubMed] [Google Scholar]
- Parkhurst CN, Yang G, Ninan I, et al. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013 ; 155 : 1596–1609. [CrossRef] [PubMed] [Google Scholar]
- Daches S, Kovacs M, George CJ, et al. Childhood adversity predicts reduced physiological flexibility during the processing of negative affect among adolescents with major depression histories. Int J Psychophysiol 2017 ; 121 : 22–28. [CrossRef] [PubMed] [Google Scholar]
- Lebe M, Hasenbring MI, Schmieder K, et al. Association of serotonin-1A and -2A receptor promoter polymorphisms with depressive symptoms, functional recovery, and pain in patients 6 months after lumbar disc surgery. Pain 2013 ; 154 : 377–384. [CrossRef] [PubMed] [Google Scholar]
- Kishi T, Yoshimura R, Ikuta T, Iwata N. Brain-derived neurotrophic factor and major depressive disorder: evidence from meta-analyses. Front Psychiatry 2017 ; 8 : 308. [CrossRef] [PubMed] [Google Scholar]
Liste des figures
|
Figure 1. Représentation schématique de la structure de l’adiponectine et de ses oligomères. La forme entiere de l’adiponectine comprend 244 acides amines, un domaine de type collagene vers l’extremite N-terminale et un domaine globulaire de type C1q a son extremite C-terminale. Dans le plasma, les monomeres d’adiponectine se combinent par leur domaine globulaire pour former des complexes multimeriques tels que des trimeres, des hexameres ou des gros multimeres de haute masse moleculaire. Il existe egalement une forme globulaire circulante de l’adiponectine. |
| Dans le texte | |
|
Figure 2. Représentation schématique des mécanismes d’action de l’adiponectine globulaire sur la microglie. L’adiponectine globulaire, en se liant a son recepteur AdipoR1 present a la surface des cellules microgliales, stimule l’activation de l’AMPKα (AMP-activated protein kinase) en favorisant la phosphorylation du residu threonine en position 172. Ceci empeche la degradation de la kinase I-B et la translocation de la sous-unite p65 de NF-B (nuclear factor-kappa B) dans le noyau, ou elle se lie a l’ADN sur des sequences consensus presentes dans les promoteurs de nombreux genes codant des proteines pro-inflammatoires comme les cytokines IL(interleukine)-1, IL-6 et TNF- tumor necrosisfactor-). L’adiponectine limite egalement l’expression de la iNOS (inducible nitric oxide synthase) et la liberation de monoxyde d’azote. |
| Dans le texte | |
|
Figure 3. Schéma illustrant comment l’environnement enrichi pourrait lutter contre les comportements anxio-dépressifs induits par le stress. La liberation chronique dans le sang de corticosterone, hormone de stress produite par les glandes surrenales, agit au niveau cerebral en promouvant une activation pro-inflammatoire de la microglie, induisant ainsi l’etablissement d’un etat neuroinflammatoire et l’apparition de comportements anxio-depressifs. A l’inverse, des conditions de vie favorables ou se melent activites physiques, sociales et cognitives, telles que celles fournies par l’environnement enrichi, permettent l’augmentation des taux d’adiponectine, adipokine produite par le tissu adipeux, qui va promouvoir dans le cerveau un phenotype microglial antiinflammatoire et ainsi lutter contre la neuroinflammation et les comportements anxio-depressifs. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.