")
")
| Issue |
Med Sci (Paris)
Volume 33, Number 11, Novembre 2017
|
|
|---|---|---|
| Page(s) | 929 - 932 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/20173311005 | |
| Published online | 04 December 2017 | |
Vers de nouveaux marqueurs pronostiques dans les leucémies aiguës myéloïdes
Analyse génomique des altérations du nombre de copies
Copy-number analysis identifies new prognostic marker in acute myeloid leukemia
Univ. Lille, Inserm, CHU Lille, UMR-S 1172 - JPArc-Centre de Recherche Jean-Pierre Aubert Neurosciences et Cancer, F-59000 Lille, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
La leucémie aiguë myéloïde (LAM) est une hémopathie maligne caractérisée par une prolifération anarchique de précurseurs myéloïdes, appelés blastes. Il s’agit d’une entité très hétérogène, sur le plan clinique d’abord, avec une grande variabilité inter-individuelle des symptômes initiaux et de la réponse aux thérapeutiques. La survie à 5 ans varie de 10 % chez les sujets âgés à 60 % chez l’enfant et le jeune adulte [1]. L’hétérogénéité se manifeste également au niveau biologique, avec huit sous-types cytologiques et des caractéristiques immuno-phénotypiques, cytogénétiques et moléculaires différentes selon les patients [2]. Malgré cette hétérogénéité, le traitement par chimiothérapie, à base d’anthracyclines et de cytarabine, reste identique chez tous les patients. Il est souvent complété par une allogreffe de cellules souches hématopoïétiques (CSH).
Actuellement, la prise en charge thérapeutique, fondée sur une stratification pronostique en trois groupes de risque (favorable, défavorable ou intermédiaire, ce dernier regroupant les 2/3 des patients), repose presque exclusivement sur les anomalies cytogénétiques [2]. Cette classification reste cependant imparfaite puisque la réponse aux traitements et le pronostic varient au sein d’un même groupe de risque. Afin d’améliorer la prise en charge des patients, il apparaît essentiel de trouver de nouveaux marqueurs pronostiques. Dans cette optique, notre équipe s’est intéressée aux altérations du nombre de copies (CNA, copy number alteration) comme potentiels marqueurs pronostiques [3].
Les cellules cancéreuses sont caractérisées par l’accumulation et la combinaison de multiples anomalies génétiques et épigénétiques [4, 5] (→).
(→) Voir la Synthèse de F. Damm et al., m/s n° 5, mai 2012, page 449
Celles-ci pourraient constituer des marqueurs pronostiques facilement identifiables, comme c’est le cas pour la délétion du locus IKZF1 (IKAROS family zinc finger 1) dans les leucémies aiguës lymphoblastiques B de l’enfant [6], le cancer de la prostate [7] ou les tumeurs solides pédiatriques [8]. Cette approche serait particulièrement intéressante en cas d’échec des diagnostics par caryotypage (qui concerne 10 % des patients).
À l’heure actuelle, même si certaines études ont montré un impact entre présence de CNA, au sens large, et un pronostic défavorable dans les LAM [9], aucune CNA spécifique n’a été mise en évidence.
À partir d’échantillons de moelle osseuse provenant de 119 patients atteints de LAM issus de deux centres français (ceux de Lille et Paris), appariés au temps du diagnostic et au moment de la rémission complète, la technologie SNP (single nucleotide polymorphism)-array pangénomique haute résolution [10] ( Figure 1 ) a été utilisée afin de mettre en évidence des altérations du nombre de copies (CNA) potentiellement pronostiques.
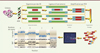 |
Figure 1. La technique de SNP-array. Cette technique permet de déterminer le variant de séquence (single nucleotide polymorphism) et le nombre de copies d’une région génétique. L’ADN d’un patient est isolé à partir des cellules sanguines ; les deux brins (allèles) sont ensuite digérés par des enzymes de restriction (ER), puis amplifiés par PCR (polymerase chain reaction) et regroupés. Des sondes marquées par des fluorochromes sont hybridées sur les puces « SNP-array » du génome humain. La quantification du signal d’hybridation de chaque sonde se fait par une lecture avec un laser ; aboutissant après numérisation à une quantification du nombre de copies pour chaque région étudiée (exprimée sous forme d’un rapport logarithmique). Cette technique offre l’avantage d’une haute résolution (identification de zones < 150 kb) et elle permet une analyse pangénomique (figure modifiée d’après Alkan et al. [10]). |
Les CNA identifiées comme étant associées à la réponse au traitement, ou au pronostic, ont ensuite été étudiées dans une cohorte nationale indépendante composée de 248 patients (dite cohorte de validation), ainsi que sur 170 échantillons provenant de l’atlas du génome du cancer (TCGA pour the cancer genome atlas), afin d’étudier leur spécificité.
L’analyse pan-génomique des CNA permet l’identification d’un nouveau marqueur pronostique indépendant dans les LAM
Des altérations du nombre de copies (CNA) ont été retrouvées dans environ 50 % des échantillons de LAM. De façon prédominante, les patients atteints de LAM présentaient une CNA unique : les délétions étaient deux à trois fois plus fréquentes que les amplifications et certains chromosomes étaient préférentiellement touchés (chromosomes 8, 11 et 21 pour les amplifications, et 7, 12, 17 et 21 pour les délétions). Il n’a pas été retrouvé d’association entre les CNA et la classification ELN (European leukemia net) actuelle. Il était donc supposé que la présence d’une ou plusieurs CNA, n’était pas, à elle seule, un marqueur pronostique, et que seules certaines CNA seraient associées au pronostic. Quatre CNA ont été identifiées pour être associées au pronostic : trois amplifications (2 sur le chromosome 21, et une sur le chromosome 11) et une délétion (sur le chromosome 11). La présence d’une de ces 4 CNA (définie comme « critère CNA ») augmente la mortalité de 4 à 5 fois avec une survie moyenne à 1,6 an par rapport aux 5 ans observés chez les patients non porteurs de ce critère. Ces résultats ont été confirmés dans la cohorte de validation, de manière indépendante, ainsi que dans la cohorte TCGA.
Par des analyses multivariées, après ajustement sur les facteurs pronostiques de la classification ELN actuelle, il a été confirmé que le nouveau « critère CNA » était corrélé à une moins bonne survie globale.
L’analyse du profil d’expression génomique des CNA identifie le gain du gène ERG comme un marqueur pronostique principal
Les quatre CNA identifiées impliquent 26 gènes aux fonctions biologiques diverses. Afin de caractériser les gènes potentiellement impliqués dans la réponse au traitement, les taux d’ARNm (ARN messagers) exprimés par les cellules ont été étudiés. Dans la cohorte TCGA, l’amplification de la région 21q22.2, contenant le gène ERG (ETS [erythroblast transformation-specific]-related gene), apparaît associée significativement à une augmentation de l’expression de l’ARNm ERG. Une association entre diminution de la survie globale et gain d’ERG est également observée dans les deux cohortes indépendantes, et dans la cohorte TCGA. Il semblerait donc que le gène ERG, un oncogène déjà connu pour son implication dans le cancer prostatique, le sarcome d’Ewing et les leucémies aiguës [11], soit un critère de mauvais pronostique.
L’association entre mutations déjà identifiées dans les LAM et « critère CNA » identifie la mutation du gène TP53 comme un marqueur pronostic, dans le groupe de risque défavorable
Il n’a pas été retrouvé d’association entre le « critère CNA » et les mutations couramment décrites dans les LAM comme celles affectant les gènes IDH1/2 (isocitrate déshydrogénases), DNMT3A (DNA méthyltransférase), RUNX1 (Runt-related transcription factor), TET2 (Tet methylcytosine dioxygenase 2), ASXL1 (additional sex comb-like 1), NPM1 (Nucleophosmin 1), FLT3 (FMS-like tyrosine kinase 3), CEBPα (CCAAT/enhancer-binding protein alpha), MLL-PTD (Mixed lineage leukaemia gene-partial tandem duplications), ou une surexpression d’EVI1 (ecotropic virus integration site 1 transcription factor). Cependant, le « critère CNA » est fréquemment associé avec une mutation de TP53 (le gène qui code p53, un suppresseur de tumeur), cette dernière étant l’un des critères ELN du groupe de risque défavorable. La mutation de TP53 est de mauvais pronostic, sur les deux cohortes étudiées et également sur la cohorte TCGA. Les patients porteurs d’une mutation de TP53 ont également un nombre médian de CNA élevé (8,5 versus 1, chez les patients porteurs du gène TP53 non muté) suggérant une plus grande instabilité génétique induite par cette mutation dans les blastes.
Un variant de TP53 a été retrouvé chez 71 % des patients qui présentent un gain d’ERG. Une analyse multivariée a montré que le « critère CNA » et le gain d’ERG seraient plus forts que les critères actuellement impliqués dans la classification ELN, dont le statut mutationnel du gène TP53. Les patients du groupe intermédiaire, ayant un « critère CNA » avec un gain d’ERG, devraient donc être inclus dans le groupe de risque défavorable.
Découverte de nouvelles sous-classes pronostiques dans la LAM
En comparant la valeur pronostique des critères de la classification ELN ( Figure 2 ) avec les facteurs mis en avant dans l’étude (nombre de CNA, « critère CNA », gain d’ERG, surexpression d’ERG et mutation de TP53), le « critère CNA » et le gain d’ERG ont donc été identifiés, comme les deux marqueurs prédictifs péjoratifs les plus forts. L’impact de ce nouveau marqueur (le critère CNA) a donc été testé dans la classification actuelle sur la cohorte de validation et la cohorte TCGA en créant deux groupes de risque supplémentaires : un groupe « très défavorable » (sous-groupe du groupe de risque défavorable) regroupant les patients ayant un « critère CNA » ou une mutation de TP53 et un groupe « défavorable-like » (sous-groupe du groupe de risque intermédiaire) ayant un « critère CNA ». Tous les patients du groupe très défavorable avaient une survie globale de moins de 2 ans. La survie était moins bonne dans le groupe « défavorable-like » que dans le groupe intermédiaire, mais la différence ne s’est pas avérée significative. Dans ces deux cohortes, la création de ces deux nouvelles sous classes permettraient de reclasser respectivement 15 et 19 % des patients.
 |
Figure 2. Classification des patients atteints de LAM. Classification ELN (European leukemia net) actuelle et proposition d’une nouvelle classification pour la stratification thérapeutique des patients atteints de LAM. ASXL1 : additional sex comb-like 1 ; BCR-ABL : fusion of breakpoint cluster region and Abelson tyrosine-protein kinase 1 ; CEBPA : CCAAT/enhancer-binding protein A ; DEK-NUP : fusion of the DEK oncogene and the nucleoporin ; EV1 : ecotropic virus integration site 1 transcription factor ; FLT3 : FMS-like tyrosine kinase 3 ; GATA : family of zinc-finger transcription factors ; KMT2A : lysine methyltransferase 2A ; MLLT3 : Mixed lineage leukaemia ; NPM1 : Nucleophosmin 1 ; RUNX1 : Runt-related transcription factor ; BAF : B allele frequency (figure établie d’après Nibourel et al. [3] et Döhner et al. [12]). |
Association entre le « critère CNA » et la chimiorésistance
Afin de mieux comprendre le mécanisme impliqué dans l’impact pronostique du « critère CNA », il a été déterminé si celui-ci pouvait être lié à la maladie réfractaire aux traitements. Dans le groupe de patients avec le « critère CNA », la fréquence de la maladie réfractaire est plus élevée que dans le groupe sans « critère CNA » (41 versus 14 %). En particulier, la présence d’un gain d’ERG dans la région 21q22.2 a été détectée dans 80 % des LAM réfractaires avec un « critère CNA ». La maladie réfractaire étant liée à la chimiorésistance, cette piste a été étudiée. Des études complémentaires de chimiorésistance in vitro ont mis en évidence une plus grande chimiorésistance à la cytarabine (le principal agent cytotoxique utilisé dans les LAM) en lien avec la surexpression d’ERG. Ceci signifie que l’utilisation de thérapies alternatives, remplaçant la cyarabine pourrait être bénéfique dans ce sous-groupe de patients.
En conclusion, deux nouveaux marqueurs pronostiques sont décrits dans cette étude : le « critère CNA » et le gain d’ERG, constituant deux marqueurs spécifiques, robustes et universels de mauvais pronostic dans les LAM ; auxquels s’ajoute la mutation de TP53, déjà connue et comprise dans la classification [12]. Ces résultats permettent de proposer une nouvelle classification pour la stratification thérapeutique des patients atteints de LAM ( Figure 2 ). À l’avenir, si cette nouvelle classification est validée de façon prospective, elle pourrait permettre une prise en charge plus adaptée à chaque patient et une amélioration du pronostic.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Références
- Maynadié M, Angelis RD, Marcos-Gragera R, et al. Survival of European patients diagnosed with myeloid malignancies: a HAEMACARE study. Haematologica 2013 ; 98 : 230–238. [CrossRef] [PubMed] [Google Scholar]
- Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 2009 ; 114 : 937–951. [Google Scholar]
- Nibourel O, Guihard S, Roumier C, et al. Copy-number analysis identified new prognostic marker in acute myeloid leukemia. Leukemia 2017 ; 31 : 555–564. [CrossRef] [PubMed] [Google Scholar]
- Beroukhim R, Mermel CH, Porter D, et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010 ; 463 : 899–905. [CrossRef] [PubMed] [Google Scholar]
- Damm F, Ngyuen-Khac F, Kosmider O, et al. Mutations des gènes impliqués dans l’épissage dans les hémopathies malignes humaines. Med Sci (Paris) 2012 ; 28 : 449–453. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Mullighan CG, Su X, Zhang J, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med 2009 ; 360 : 470–480. [Google Scholar]
- Hieronymus H, Schultz N, Gopalan A, et al. Copy number alteration burden predicts prostate cancer relapse. Proc Natl Acad Sci USA 2014 ; 111 : 11139–11144. [CrossRef] [Google Scholar]
- Harris MH, DuBois SG, Glade Bender JL, et al. Multicenter feasibility study of tumor molecular profiling to inform therapeutic decisions in advanced pediatric solid tumors: the individualized cancer therapy (iCat) study. JAMA Oncol 2016; Jan 28. doi: 10.1001/jamaoncol.2015.5689. [Google Scholar]
- Parkin B, Ouillette P, Yildiz M, et al. Integrated genomic profiling, therapy response, and survival in adult acute myelogenous leukemia. Clin Cancer Res 2015 ; 21 : 2045–2056. [CrossRef] [PubMed] [Google Scholar]
- Alkan C, Coe BP, Eichler EE. Genome structural variation discovery and genotyping. Nat Rev Genet 2011 ; 12 : 363–376. [CrossRef] [PubMed] [Google Scholar]
- Martens JHA. Acute myeloid leukemia: a central role for the ETS factor ERG. Int J Biochem Cell Biol 2011 ; 43 : 1413–1416. [CrossRef] [PubMed] [Google Scholar]
- Döhner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017 ; 129 : 424–447. [Google Scholar]
© 2017 médecine/sciences – Inserm
Liste des figures
|
Figure 1. La technique de SNP-array. Cette technique permet de déterminer le variant de séquence (single nucleotide polymorphism) et le nombre de copies d’une région génétique. L’ADN d’un patient est isolé à partir des cellules sanguines ; les deux brins (allèles) sont ensuite digérés par des enzymes de restriction (ER), puis amplifiés par PCR (polymerase chain reaction) et regroupés. Des sondes marquées par des fluorochromes sont hybridées sur les puces « SNP-array » du génome humain. La quantification du signal d’hybridation de chaque sonde se fait par une lecture avec un laser ; aboutissant après numérisation à une quantification du nombre de copies pour chaque région étudiée (exprimée sous forme d’un rapport logarithmique). Cette technique offre l’avantage d’une haute résolution (identification de zones < 150 kb) et elle permet une analyse pangénomique (figure modifiée d’après Alkan et al. [10]). |
| Dans le texte | |
|
Figure 2. Classification des patients atteints de LAM. Classification ELN (European leukemia net) actuelle et proposition d’une nouvelle classification pour la stratification thérapeutique des patients atteints de LAM. ASXL1 : additional sex comb-like 1 ; BCR-ABL : fusion of breakpoint cluster region and Abelson tyrosine-protein kinase 1 ; CEBPA : CCAAT/enhancer-binding protein A ; DEK-NUP : fusion of the DEK oncogene and the nucleoporin ; EV1 : ecotropic virus integration site 1 transcription factor ; FLT3 : FMS-like tyrosine kinase 3 ; GATA : family of zinc-finger transcription factors ; KMT2A : lysine methyltransferase 2A ; MLLT3 : Mixed lineage leukaemia ; NPM1 : Nucleophosmin 1 ; RUNX1 : Runt-related transcription factor ; BAF : B allele frequency (figure établie d’après Nibourel et al. [3] et Döhner et al. [12]). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.