")
")
| Issue |
Med Sci (Paris)
Volume 25, Number 12, Décembre 2009
Anticorps monoclonaux en thérapeutique
|
|
|---|---|---|
| Page(s) | 1024 - 1032 | |
| Section | I - De la conception à la production | |
| DOI | https://doi.org/10.1051/medsci/200925121024 | |
| Published online | 15 December 2009 | |
Anticorps thérapeutiques et dérivés : une palette de structures pour une pléthore d’indications
Quel format et quelle glycosylation choisir ? Pour quelles applications ?
Therapeutic antibodies and related products: choosing the right structure for success
Centre d’immunologie Pierre Fabre (www.cipf.com), 5, avenue Napoléon III, 74160 Saint-Julien-en-Genevois, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
Les anticorps monoclonaux (Acm) et dérivés constituent la classe de principes actifs qui connaît le plus fort taux de développement actuel dans les domaines biotechnologique et pharmaceutique. En un peu plus de vingt ans, plus de 30 immunoglobulines (IgG) et dérivés ont obtenu une autorisation de mise sur le marché (AMM) dans des indications thérapeutiques très variées (oncologie, inflammation et auto-immunité, transplantation, angioplastie, hématologie, ophtalmologie, infections virales, allergie). La palette de structures comporte des anticorps murins, chimériques, humanisés et humains d’isotypes différents (IgG1, 2 et 4), ainsi que des dérivés d’IgG (immunoconjugués, radio-immunoconjugués, fragments Fab, protéines et peptides de fusion Fc, bispécifiques souris/rat). De plus, du point de vue structural, leur glycosylation dépend du système de production et peut influencer les fonctions effectrices et l’immunogénicité. À partir des connaissances actuelles des relations structure-fonction, quels formats d’anticorps est-il préférable de choisir ? Avec quels types de glycosylation ? Pour quelles indications ?
Abstract
Monoclonal antibodies (mAb) and related-products represent the fastest growing class of therapeutics in the biotechnological and pharmaceutical industry. In just as short as 20 years, more than 30 immunoglobulins (IgG) and derivatives have been approved in a wide range of indications (oncology, inflammation and auto-immunity, transplantation, angioplasty, hematology, ophthalmology, viral infections, allergy). The mAb structure toolbox contains mouse, chimeric, humanized and human antibodies from different isotypes (IgG1, 2 and 4), as well as IgG-related products (immunoconjugates, radio-immunoconjugates, Fab fragments, Fc-fusion proteins and peptides, bispecifics). Furthermore from a structural point of view, mAb glycosylation is linked to their production systems and may impact on their effector functions and immunogenicity. Based on the current knowledge, choosing the right antibody format, isotype and glycosylation profile are some of the key issues to address early during the lead selection.
© 2009 médecine/sciences - Inserm / SRMS
Le succès médical des anticorps monoclonaux (Acm) est lié à leurs caractéristiques structurales et fonctionnelles : forte affinité (de l’ordre du nM ou pM) et très grande spécificité pour leur antigène (Ag), diversité des mécanismes d’action (antagonisme/agonisme, signalisation, internalisation/dégradation de l’Ag, induction d’apoptose, fonctions effectrices cytotoxiques, etc.), pharmacocinétique modulable selon le format (de quelques heures à 21 jours) (→), faible toxicité intrinsèque (leur structure est similaire à celle des immunoglobulines (Ig) endogènes, et ils sont métabolisés en acides aminés (aa) naturels [1].
(→) voir G. Paintaud, page 1057
Le succès pharmaceutique et industriel des Acm s’explique par la maturité technologique qui permet de produire des Acm sous forme recombinante dans des cellules de mammifères [2]. Il est ainsi possible de passer en 9 à 12 mois de lots de type « recherche » (bioréacteur de 3 l) à des lots utilisables dans des essais cliniques de phase I (300 à 2 000 l), dans un environnement de bonnes pratiques de fabrication (GMP, good manufacturing practices) [3], puis d’étendre la production de 5 à 10 000 l pour des essais de phases II/III et jusqu’à 20 000 l après obtention de l’autorisation de mise sur le marché (AMM) (→). Parallèlement, la très grande homologie de structure des IgG (> 90 %) permet la mise en place de plateformes d’ingénierie, de purification, de formulation et de caractérisation analytique [4, 5] favorisant la recherche et le développement de plus en plus rapides de ces biomolécules.
(→) voir O. Cochet et M. Chartrain, page 1078
Toutes ces caractéristiques contribuent à un taux d’attrition1 en clinique plus faible pour les Acm que pour les petites molécules chimiques [6]. Ces dernières conservent l’avantage d’une administration per os, de la possibilité d’atteindre des cibles intracellulaires et de pouvoir être produites pour un prix de revient industriel plus bas.
Le Tableau I liste par catégories les Acm et dérivés qui ont été approuvés pour un usage thérapeutique par la FDA (Food and drug administration), l’EMEA (European medicines agency) ou la SFDA (China state food and drug administration).
Anticorps monoclonaux et dérivés approuvés pour un usage thérapeutique (FDA, EMEA ou SFDA). * DCI = dénomination commune internationale (www.who.org). Elle est composée par un préfixe unique, suivi d’un infixe représentant la pathologie ciblée (tu = tumeur ; cir = cardiovasculaire ; vi = virus ; lim = immune) et un suffixe lié au type d’AcM [souris = -omab ; chimère = -ximab (souris, rat, primate non humain) ; humanisé = -zumab ; humain = -umab]. La dernière consonne de la syllabe de la cible/ maladie peut être éliminée pour faciliter la prononciation (ex. palivizumab, trastuzumab, rituximab, etc.). Le suffixe des protéines-Fc est -cept ; celui des peptibody -stim. ** Année du 1er enregistrement FDA, EMEA ou SFDA. *** À titre d’exemple, les structures de ces Acm et dérivés sont schématisées sur les Figures 2 et 3. **** Les valeurs des demi-vies et d’immunogénicité sont données à titre indicatif (sources = publications scientifiques, sites web EMEA, FDA et des laboratoires). Elles peuvent varier en fonction des antigènes, des méthodes de dosages, des populations incluses dans les études cliniques et d’autres paramètres (→).
(→) voir G. Paintaud, page 1057 ; L. Manache et al., page 1063 ; P. Stas et I. Lasters, page 1070
Abréviations : CCR = cancer colorectal ; DMLA = dégénérescence maculaire liée à l’âge ; EMEA = European Medicines Agency ; EGFR : epidermal growth factor receptor ; epCAM : epithelial cell adhesion molecule ; FDA = Food and Drug Administration ; HPN = hémoglobinurie paroxystique nocturne ; i.m. = intramusculaire ; i.o. = intraoculaire ; i.p. : intrapéritonéal ; i.v. = intraveineux ; IC = immunoconjugué ; LAM = leucémie aiguë myéloïde ; LLC = leucémie lymphocytaire chronique ; LNH = lymphome non hodgkinien ; PR = polyarthrite rhumatoïde ; PTI = purpura thrombopénique idiopathique ; RIC = radioimmunonoconjugué ; s.c. = sous-cutané ; SA = spondylarthrite ankylosante ; SFDA = China State Food and Drug Administration ; SPAC = syndromes périodiques associés à la cryopyrine ; TNFalpha : tumor necrosis factor alpha ; TPO-R : récepteur de la thrombopoïétine ; VEGF : vascular endothelial growth factor ; NSO, CHO, SP2/20 : lignées cellulaires utilisées pour la production.
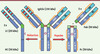 |
Figure 1. hIgG1, chaînes lourde et légère, fragments Fab et Fc. Les région variables sont représentées en bleu clair, les régions constantes en bleu foncé, les CDR en rouge, la région charnière en vert, les ponts disulfure inter-chaînes en brun, les ponts disulfure intra-chaînes en orange et les glycanes en rose. Chaînes lourde (H), légère (L), variable (V), constante (C). |
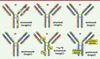 |
Figure 2. Exemples d’IgG murines, chimériques, humanisées et humaines (nues et conjuguées). Les exemples illustrés ont été approuvés pour un usage thérapeutique. Les régions variables murines sont représentées en gris clair, les régions constantes murines en gris foncé, les régions variables humaines sont représentées en bleu clair, les régions constantes humaines en bleu foncé, les CDR murins en rouge et humains en bleu foncé, la région charnière murine en vert foncé et humaine en vert clair, les ponts disulfure en orange et les glycanes en rose. |
 |
Figure 3. Exemples de fragments d’Ig approuvés pour un usage thérapeutique (Fab, Fab-pegylé, Fc-protéine et peptide-Fc). Les régions variables humaines sont représentées en bleu clair, les régions constantes en bleu foncé, les CDR en rouge, la région charnière en vert, les résidus PEG en jaune, les ponts disulfure en orange, la partie extracellulaire du récepteur au TNF en bleu et les glycanes en rose. |
Les immunoglobulines humaines réparties en cinq classes (IgA, IgD, IgE, IgG et IgM) sont formées de deux types de chaînes polypeptidiques, qualifiées de légères (IG L- L pour light, deux sous-classes : κ et λ) et lourdes (IG H - H pour heavy, neuf sous-classes ou isotypes : α1 et 2 ; δ ; ε ; γ1, 2, 3 et 4 ; µ). Les classes et les sous-classes sont définies par la nature de leurs chaînes lourdes qui conditionne des différences de structure, de fonctionnalité et de fonction dans la défense immunitaire. À l’heure actuelle, tous les anticorps thérapeutiques et leurs dérivés ayant obtenu une AMM sont des IgG, et la majorité sont humanisées (voir ci-dessous) (→). Seules quelques phases cliniques précoces ont été menées pour l’instant avec des IgA et des IgM. Celles-ci présentent des structures oligomériques (dimères, pentamères) et une glycosylation (N et O) beaucoup plus complexes que celles des IgG.
(→) voir M. Cogné et al., page 1149
Immunoglobulines murines et humaines : structures et fonctions
Les IgG murines et humaines sont des glycoprotéines tétramériques bivalentes (deux sites de liaison aux Ag) et bifonctionnelles (liaison aux Ag et aux γ-Fc récepteurs) (Figure 1). Ce sont des hétérodimères d’environ 150 kDa constitués de deux chaînes lourdes (50 kDa ≈ 450 aa) et de deux chaînes légères (25 kDa ≈ 220 aa). Ces chaînes polypeptidiques sont liées par un nombre caractéristique de ponts disulfures (S-S), impliqués dans des appariements particuliers à chaque isotype. Les chaînes légères sont constituées d’un domaine variable (VL) et d’un domaine constant (CL) ; les chaînes lourdes d’un domaine variable (VH), de trois domaines constants (CH1, 2 et 3) et d’une région charnière (H, hinge), située entre les domaines CH1 et CH2. Les chaînes lourdes comprennent un site conservé de N-glycosylation situé généralement sur l’Asn297 [7] (l’Asn84.4 du domaine CH2 dans la numérotation IMGT2 (→). Les domaines variables des chaînes lourdes et légères comportent chacun trois régions hypervariables impliquées dans la spécificité de reconnaissance de l’Ag (CDR, pour complementarity determining regions ou région déterminant la complémentarité avec l’antigène) en alternance avec des domaines charpentes (FR pour framework). La charnière, très flexible, comprend les ponts S-S qui relient les chaînes lourdes entre elles. Ce domaine comporte un site de clivage enzymatique privilégié (papaïne) qui permet de fragmenter l’Acm en Fab (fragment se liant à l’Ag) et Fc (fragment cristallisable), d’environ 50 kDa chacun. Le Fab est responsable de la liaison spécifique de forte affinité aux Ag. Le fragment Fc se lie à des γ-Fc récepteurs impliqués dans les fonctions effectrices des Acm (ADCC = cytotoxicité cellulaire dépendante d’anticorps, CDC = cytotoxicité dépendante du complément), ainsi qu’à des FcRn qui confèrent une demi-vie plasmatique d’une vingtaine de jours aux IgG humaines (→), excepté les IgG3 où elle est de 7 jours.
(→) voir M.P. Lefranc, page 1020
(→) voir R. Abès et al., p. 1011 ; C. Magdelaine-Beuzelin et al., page 1053 ; G. Paintaud, page 1057
Immunoglobulines murines, chimériques (souris, rat, singe), humanisées et humaines
L’idée de Paul Ehrlich formulée en 1900 d’utiliser la spécificité des anticorps pour neutraliser un agent pathogène [1] s’est concrétisée en 1975 par la mise au point de la production d’Acm murins via la technologie des hybridomes par Georges Köhler et César Milstein, ce qui leur valut le prix Nobel en 1984 [8] (→). Onze ans plus tard, muromomab (Orthoclone, anti-CD3) (Figure 2A) fut le premier Acm à être approuvé pour lutter contre les rejets de greffe (→). L’utilisation d’Acm de souris chez l’homme a cependant rapidement été limitée par la forte immunogénicité de ces molécules et leur demie-vie courte chez l’homme (1 jour).
(→) voir H. Watier, page 999
(→) voir B. Vanhove, page 1121
Une amélioration significative consista en 1984 à produire des Acm chimériques constitués par des domaines variables de souris fusionnés à des domaines constants humains [9]. Dix ans plus tard, abciximab (Réopro®, dirigé contre la glycoprotéine plaquettaire GPIIb/IIIa) fut le premier Acm chimérique approuvé sous un format de fragment Fab. Il a été suivi de rituximab (Rituxan/Mabthéra®, anti-CD20), sous un format d’IgG entière. Un autre saut technologique consista en 1986 à humaniser les IgG en greffant des CDR de souris sur des charpentes humaines [10]. Daclizumab (Zenapax, anti-CD25) fut le premier Acm de cette classe à être approuvé, il fut suivi de nombreux autres. Finalement, deux autres technologies permettant de générer des Acm humains furent proposées en 1990 [11] et 1994 [12, 13]. La première était fondée sur des banques de phages3 reflétant le répertoire naturel des lymphocytes B humains, la seconde sur des souris transgéniques dont le système immunitaire avait été modifié de façon à générer des anticorps humains en réponse à une immunisation par un Ag d’intérêt [14] (→). Les deux approches ont démontré leur efficacité comme en témoigne la mise sur le marché respectivement en 2002 d’adalimumab (Humira®, anti-TNFα, tumor necrosis factor) et en 2006 de panitumumab (Vectibix®, anti-EGFR, epidermal growth factor receptor).
(→) voir M. Cogné et al., page 1149
À l’heure actuelle, les IgG humanisées représentent la majorité des Acm présents sur le marché et en développement. De nombreux Acm humains (plus de 66) issus de banques de phages ou de souris transgéniques sont également en développement [15]. L’accès aux banques de phages (Dyax, CAT, Cambridge antibody technology, absorbé par Astra-Zeneca) et aux souris transgéniques (Medarex, absorbé par Bristol-Myers Squibb, et Abgenix absorbé par Amgen) est couvert par de nombreux brevets et n’est accessible généralement qu’à de grandes sociétés pharmaceutiques qui ont tendance à acquérir les sociétés détentrices des technologies clés. De petites sociétés de biotechnologie continuent d’utiliser le format chimérique pour éviter de verser des redevances liées à l’humanisation. BiogenIdec développe plusieurs Acm « primatisés » chimériques singe/humain (lumiximab, anti-CD23, testé dans les leucémies lymphoïdes chroniques ; galiximab, anti-CD80, testé dans les lymphomes non hodgkiniens), actuellement en phases tardives d’essais cliniques. Le format murin a été abandonné, sauf dans le cas de certains radio-immunoconjugués où une demi-vie très courte est recherchée pour limiter la radiotoxicité (→). Une exception notable est l’autorisation de mise sur le marché en Europe en avril 2009 du catumaxomab, un anticorps bi-fonctionnel constitué par des chaînes légère et lourde de souris (IgG2a, anti-EpCAM, epithelial cell adhesion molecule [45]), combinées à des chaînes légère et lourde de rat (IgG2b, anti-CD3) et produit par une technologie de type quadroma4. Son utilisation est à l’essai dans les ascites métastatiques.
(→) voir J. Barbet et al., page 1039
Structure des IgG : choix de l’isotype IgG1, IgG2 ou IgG4 ?
Les IgG1 (ex. daclizumab, Figure 2C) et IgG4 (ex. gemtuzumab, Figure 2E) possèdent seize ponts S-S et les IgG2 deux de plus (ex. panitumumab, Figure 2D). Les IgG1 et les IgG4 sont caractérisées par deux ponts interchaînes lourdes au niveau de la charnière mais se différencient par un appariement différent de chaînes lourde-légère. Les IgG2 ont quatre ponts interchaînes lourdes, qui les rendent plus rigides, et un appariement chaînes lourde-légère similaire à celui des IgG4. Les IgG3 possèdent une région charnière beaucoup plus longue (onze ponts S-S). Les autres Cys sont impliquées dans des ponts intramoléculaires dans chaque domaine globulaire (VL, CL, VH, CH1, CH2 et CH3).
Les IgG sont présentes dans le plasma humain à une concentration de 10 g/L et avec un rapport entre les sous-classes 66/23/7/4 (IgG1/2/3/4). La plupart des IgG chimériques, humanisées et humaines approuvées sont des IgG1 (rituximab, trastuzumab, palivizumab, adalimumab, etc.). Ces Acm sont capables à la fois de se lier à leur Ag et, lorsque celui-ci est localisé à la surface membranaire, de détruire les cellules associées grâce à des fonctions effectrices du type ADCC (antibody dependent cellular cytotoxicity) ou CDC (complement dependent cytotoxicity) [16] (→). Ce format est souvent choisi en oncologie lorsque l’objectif est à la fois de cibler et de détruire les cellules tumorales [17].
(→) voir R. Abès et al., page 1011
Inversement, les isotypes IgG4 et 2 sont en général choisis lorsque les fonctions effectrices ne sont pas souhaitées. De nouvelles données sur les relations structure-fonction publiées récemment ont montré que ces deux formats avaient un comportement « dynamique » in vitro et in vivo plus complexe que celui des IgG1 [18]. Les IgG4 peuvent contenir des taux de « demi-anticorps » importants5, ces derniers pouvant être inactifs si le mécanisme d’action de l’anticorps nécessite une bivalence. D’autre part, des demi-anticorps peuvent se combiner de manière aléatoire pour former des anticorps bispécifiques et lier des Ag différents [19]. Les IgG2, quant à elles, peuvent former plusieurs isoformes structurales (A, B, A/B, etc.) caractérisées par des appariements différents de ponts disulfures entre chaînes lourdes et légères [20], ainsi que des dimères covalents [21]. Ces appariements sont sensibles à l’environnement redox in vitro et in vivo (par ex. au taux de glutathion réduit présent sur les hématies6) [22]. Ils peuvent varier au cours du temps (compartiment cellulaire, âge de l’anticorps) et avoir un impact sur la fonctionnalité des anticorps.
Ingénierie des IgG : amplifier les fonctions en optimisant les structures
La connaissance fine des relations structure-fonction des Acm a conduit à la construction d’IgG optimisées dont certaines ont atteint le marché [23]. Éculizumab (Soliris®, anti complément C5), par exemple, est une immunoglobuline hybride IgG2/4 dont la chaîne lourde est constituée des domaines CH1 et charnière d’une IgG2 et des domaines CH2 et CH3 d’une IgG4. Le but est d’empêcher la liaison aux γ-FcR et de ne pas activer le complément [24] (→). Une autre façon d’abroger totalement les fonctions effectrices des IgG4 consiste à muter l’Asn297 (84.4, notation IMGT) ou la Thr299 (85.3, notation IMGT) pour produire des IgG non glycosylées (-NST-). Inversement, des constructions hybrides IgG1/3 permettent d’augmenter la cytotoxicité [25]. Dans le cas des IgG4, pour éviter la formation de demi-anticorps, la Ser228 (10, notation IMGT) de la région charnière (-CPSC-) peut être remplacée par une Pro, présente dans les IgG1 (-CPPC-) [26]. L’ingénierie des CDR a été utilisée avec succès pour augmenter l’affinité pour le VEGF-A (vascular endothelial growth factor) de ranibizumab par rapport au bévacizumab ; celle des domaines charnière pour transformer un antagoniste en un agoniste [27] ; celle de la partie Fc pour augmenter ou réduire les fonctions effectrices (liaison aux γ-FcR) ou encore pour augmenter la demi-vie plasmatique (liaison aux FcRn). Par ailleurs, une multitude de nouveaux formats de taille minimaliste sont en développement dont une dizaine ont atteint un stade clinique précoce [28] (→).
(→) voir G. Socié et al., page 1126
(→) voir P. Chames et D. Baty, page 1159 ; T. Wurch et al., page 1169
Glycosylation : quelles glycoformes choisir ? Pour quelles fonctions ?
Les glycanes ne représentent que 2 % à 3 % de la masse d’un anticorps mais jouent un rôle essentiel dans les fonctions effectrices (ADCC et CDC). Ces hydrates de carbone sont localisés sur l’Asn297 (84.4, IMGT) des chaînes lourdes des IgG humaines et murines, au niveau d’un site de N-glycosylation consensus -Asn-X-Thr- (X ≠ Pro). Environ 15 % à 20 % des IgG plasmatiques possèdent un second site de N-glycosylation dans le domaine variable. C’est le cas de cetuximab (-Asn88-Asp-Thr-) (Figure 2B) [29].
Les N-glycoformes des Fc sont composées d’un heptasaccharide commun composé de deux N-acétylglucosamines (N-Glc), de trois mannoses (Man) formant deux bras liés à deux N-acétylglucosamines et à deux mannoses (Figure 4). Cet heptasaccharide est généralement complété par un fucose-1,6 (Fuc) au niveau du premier GlcNac et de 0,1 ou 2 unités de galactose (Gal) formant ainsi les trois glycoformes appelées respectivement G0F, G1F et G2F (Figure 4A). Ces trois glycoformes sont également celles que l’on retrouve majoritairement (> 80 %) sur les Fc des IgG produites dans des cellules de mammifères (CHO, NS0, SP2/0, HEK-293 ou PER.C6) [4]. Les Acm produits en systèmes murins (NS0, SP2/0) possèdent des glycoformes supplémentaires en faible quantité (< 5 %) qui peuvent être immunogéniques (Gal-α-1,3-Gal ; acide N-glycolyl neuraminique, Figure 4B) [31]. Ces glycoformes « murines », présentes à l’état de trace dans les Fc des Acm produits dans les cellules NS0 (palivizumab) et SP2/0 (daclizumab), le sont en quantité beaucoup plus importante dans la partie Fab de cétuximab (30 %). Dans le cas de cet anticorps, elles ont été reconnues responsables de réactions anaphylactiques chez des patients qui possédaient des anticorps anti-Gal-α-1,3-Gal préexistants [29].
 |
Figure 4. Glycoformes des IgG humaines et recombinantes. |
Lorsque les Acm sont produits dans des systèmes alternatifs, d’autres glycoformes immunogéniques peuvent être présentes (fucose-1,3 et xylose-1,2 chez les végétaux par exemple) (Figure 4B) [46]. Dans ce cas, ces Acm ne peuvent pas être administrés par une voie systémique. Pour remédier à ce problème, plusieurs systèmes de production ont été modifiés génétiquement pour empêcher la formation de ces glycoformes xénobiotiques en inhibant par exemple les étapes d’addition de Xyl-1,2 et de Fuc-1,3 (Biolex, Greenovation) [7]. Par ailleurs, d’autres systèmes eucaryotes ont été développés pour augmenter la part d’ADCC (40 à 100 fois, in vitro), en empêchant l’addition de Fuc-1,6 (Kyowa Kirin) [30] ou en ajoutant un bras bisecteur (GlycArt Biotechnology AG acquis par Roche). Le patrimoine génétique de la levure a également été modifié pour humaniser la glycosylation et moduler l’ADCC (GlycoFi acquis par Merck) [32]. Il existe également des lignées qui, naturellement, produisent des glycoformes non fucosylées et qui permettent de produire des Acm ayant une activité ADCC plus forte (cellules de rat YB2/0, développées par LFB Biotechnologie ; cellules souches de canard Ebx® développées par Vivalis) (→). Une autre voie intéressante d’ingénierie de la glycosylation consiste à « saturer » les deux antennes des N-glycanes en acides sialiques afin d’accroître le profil anti-inflammatoire des IgG ou simplement du fragment Fc [33, 34].
(→) voir S. Olivier et M. Mehtali, page 1163
Immunoconjugués et radio-immunoconjugués : pour une cytotoxicité accrue et ciblée
Une autre manière d’augmenter la cytotoxicité des anticorps consiste à leur conjuguer chimiquement des radioéléments ou des molécules cytotoxiques, permettant ainsi de les cibler à des cellules tumorales. Ces technologies sont cependant plus lourdes et compliquées à mettre en œuvre et n’ont connu pour l’instant qu’un succès limité et principalement en hémato-oncologie (→). Seuls quatre radio-immunoconjugués ont été mis sur le marché à l’heure actuelle (dont deux uniquement en Chine) et un seul immunoconjugué (approuvé par la FDA mais pas par l’EMEA). Dans le cas des radio-immunoconjugués, il s’agit par exemple d’anticorps murins anti-CD20 conjugués à des radio-éléments différents : ibritumomab/Zévalin® (muIgG1, 111In pour l’imagerie et 90Y pour le traitement) et tositumomab/Bexxar® (muIgG2a, 131I) approuvés dans des indications de lymphomes non hodgkiniens (LNH). Dans le cas de l’immunoconjugué, il s’agit du gemtuzumab, une IgG4 humanisée anti-CD33 conjuguée à la toxine calichéamycine (Mylotarg), indiquée dans le traitement des leucémies aiguës myéloïdes (LAM). Des développements pharmaceutiques et cliniques récents concernant de nouveaux radio-immunoconjugués (→) et immunoconjugués (→) seront décrits de manière plus détaillée dans ce numéro.
(→) voir J. Barbet et al., page 1039
(→) voir J. Barbet et al., page 1039
(→) voir J.F. Haeuw et al., page 1046
Fragments Fab (nus ou PEGylés) : un format monovalent et dépourvu de fonctions effectrices
Le format Fab est retenu dans quelques cas particuliers où la bivalence n’est pas requise et où généralement la demi-vie courte et l’absence de fonction effectrice peuvent limiter la toxicité [35]. Abciximab (Réopro, anti-GPIIb/IIIa) a été le premier Fab à être approuvé pour lutter contre la thrombose des prothèses lors des interventions d’angioplastie. Ranibizumab (Lucentis) est un Fab dérivé du bévacizumab dirigé contre le même épitope sur le VEGF-A mais avec une plus forte affinité (→). Quatre acides aminés ont été optimisés par maturation d’affinité dans deux CDR des chaînes lourdes. Ranibizumab est utilisé par voie intra-oculaire dans la dégénérescence maculaire liée à l’âge. Il faut souligner l’utilisation hors AMM de plus en plus fréquente du bévacizumab (40 fois moins cher) pour cette indica-tion, malgré les différences structurales et l’absence d’études toxicologiques spécifiques (→). Le troisième Fab à avoir reçu une AMM est le certolizumab pegol (Cimzia, anti-TNFα) [36]. Il s’agit d’un fragment conjugué de manière spécifique à deux macromolécules de polyéthylène glycol (PEG, 40 kDa), ce qui permet d’accroître de manière très significative sa demi-vie plasmatique. D’autres nouveaux formats monovalents ayant une demi-vie accrue sont en cours de développement clinique : MetMab à un bras de Genentech et Unibody de Genmab [35]. Du point de vue industriel, ces Fab (non glycosylés) et leurs dérivés peuvent être produits dans E. coli à un coût moindre que dans des cellules mammaliennes (→).
(→) voir N. Leveziel et al., page 1105
(→) voir J. Biot et al., page 1177
(→) voir O. Cochet et M. Chartrain, page 1078
Constructions hybrides : protéines et peptides de fusion avec le fragment Fc
On a également valorisé la partie Fc des IgG en la fusionnant génétiquement à des parties extracellulaires de récepteurs, et plus récemment à des peptides. Ces constructions hybrides permettent par exemple de mimer des récepteurs dimériques (grâce aux ponts S-S de la région charnière), de les rendre solubles (pas de domaine transmembranaire), d’accroître leur demi-vie plasmatique (fixation au récepteur endothélial FcγRn) et de piéger un ligand ou une cellule impliqué(e) dans une pathologie. L’étanercept (Enbrel, TNFR-Fc, TNFα, Figure 3B), l’aléfacept (Amevive, LFA-3-Fc7, CD2), l’abatacept (Orencia, CTLA4-Fc8, CD80/CD869) et le rinolacept (Arcalyst, IL-1R-Fc, IL1) ont ainsi été approuvés dans des pathologies inflammatoires majeures (arthrite rhumatoïde, psoriasis) (→) ou « orphelines » (syndromes périodiques associés à la cryopyrine). En oncologie, une protéine de fusion VEGFR-Fc de forte affinité pour le VEGF-A est actuellement évaluée dans des essais de phase III dans le cancer du poumon (aflibercept/ AVE0005).
(→) voir L. Semerano et M.C. Boissier, page 1108 ; J. Sibilia, page 1033
La partie Fc des IgG fusionnée à des peptides a également permis d’accroître considérablement leur demi-vie plasmatique. Le romiplostim (Nplate, peptide de 41 aa fusionné à un domaine Fc, TPOR) a ainsi été récemment approuvé pour le traitement des purpuras thrombopéniques idiopathiques. Il s’agit d’un « pepticorps » agoniste du récepteur de la thrombopoïétine (TPO), cytokine qui stimule la production des plaquettes par les mégacaryocytes médullaires.
Anticorps biosimilaires : copies fidèles ou structures différentes ?
Les deux premiers biosimilaires de rituximab (Reditux, Dr Reddy’s Laboratories Ltd, 2007) et d’abciximab (Clotinab, ISU ABXIS Co, Ltd, 2008) ont fait leur apparition, respectivement en Inde et en Corée du Sud [37, 38, 47]. La complexité structurale des Acm et les multiples microvariants caractérisés par des méthodes comme la spectrométrie de masse couplée à la chromatographie liquide [5, 39, 40] montrent cependant que chaque clone et chaque procédé de fabrication produit un anticorps avec des profils caractéristiques et des différences pharmacologiques qui nécessitent des études toxicologiques et cliniques particulières [41] (→).
(→) voir F. Lackner et M.E. Behr-Gross, page 1183
Conclusion
Le choix du format est guidé par les connaissances de plus en plus approfondies des relations entre les structures et les fonctions des Acm [42]. Il est également modulé par des stratégies de marketing (premier anticorps d’une nouvelle classe ou structure de seconde génération) et par des restrictions de brevets concernant notamment l’humanisation, les banques de phages, les souris transgéniques, ou les systèmes de production.
L’exemple des cinq molécules antagonisant le TNFα approuvées montre que de multiples formats peuvent être développés avec succès contre une même cible avec des stœchiométries [43] et des mécanismes d’action qui peuvent être différents, tout comme leur positionnement thérapeutique : IgG1 chimérique (infliximab, 1998), protéine de fusion Fc (étanercept, 1998), IgG humaine (adalimumab, 2002 et golimumab, 2009) ou Fab-Pégylé (certolizumab, 2008). Dans cette classe, l’adalimumab connaît la plus forte croissance d’utilisation (35 % en 2007), mais celle-ci semble plus liée au dispositif d’administration sans aiguille qu’au format IgG lui-même [44].
Les IgG humanisées et humaines représentent les deux plus grandes catégories d’Acm en développement à l’heure actuelle, avec des profils d’immunogénicité plus faibles que ceux des anticorps chimériques (→). Les bénéfices thérapeutiques et les chiffres d’affaire en progression constante (supérieure à 1 milliard d’euros) de rituximab (anti-CD20), infliximab (anti-TNFα) et cétuximab (anti-EGFR) montrent cependant qu’il est difficile de déplacer le premier Acm d’une classe donnée, en ne changeant que le format.
(→) voir P. Stas et I. Lasters, page 1070
Du point de vue de l’isotype, les IgG1 sont les plus courantes et leur format est le moins complexe à développer avec en prime des fonctions effectrices cytotoxiques (ADCC, CDC). Les IgG4 (stabilisées dans la région charnière, non glycosylées) et les IgG2 (qui présentent une plus grande complexité en termes d’isoformes liée aux ponts disulfures) sont généralement préférées lorsque les fonctions effectrices sont susceptibles d’induire une trop forte toxicité.
De plus en plus de nouveaux formats issus de l’ingénierie moléculaire (protéines-Fc ou Fab ; CDR, domaines charnières et constants, glycanes optimisés) ou chimique (conjugués) sont développés avec succès dans leurs applications pharmaceutiques et cliniques, ce qui démontre la versatilité structurale des IgG et de leurs sous-domaines.
En termes de systèmes de production, les cellules CHO et NS0, approuvées réglementairement depuis de nombreuses années, permettent de produire des IgG avec une productivité croissante (1 à 5 g/l, certainement davantage dans le futur) et une glycosylation ad hoc (G0F, G1F et G2F similaires aux IgG humaines). Alternativement, de nouveaux systèmes de production comme des levures modifiées génétiquement (Pichia pastoris) pourront produire des anticorps avec une glycosylation plus homogène et plus fidèle encore à la glycosylation humaine [48].
Conflit D’Intérêts
Les auteurs déclarent avoir des liens durables ou permanents avec l’entreprise Institut de Recherche Pierre Fabre.
Terme utilisé en marketing, le taux d’attrition ici correspond au nombre de produits qui seront perdus entre les deux phases d’essais cliniques parce qu’ils ne satisferont pas les critères.
IMGT : International immunogenetics information system for immunoglobulins or antibodies.
Le principe du phage display est rendu possible par la maîtrise des techniques d’expression de peptides à la surface des virus de types bactériophages filamenteux. Pour cela, le gène d’une protéine de surface du phage est fusionné avec les gènes d’une banque de protéines. Ainsi, chaque phage exprime à sa surface l’une des protéines du répertoire codé par la banque. Les phages couramment utilisés sont spécifiques de Escherichia coli et leur génome est sous forme d’ADN.
Quadroma : technologie de type « hybridome d’hybridome » : fusion somatique de deux hybridomes ayant chacun une spécificité donnée. Technique utilisée pour produire des anticorps disymétriques, comprenant deux chaînes légères différentes et deux chaînes lourdes différentes.
Les IgG4 peuvent se trouver sous une forme monovalente (une chaîne légère et une chaîne lourde) et de ce fait sont susceptibles de se recombiner avec d’autres anticorps pour former les anticorps naturels bispécifiques.
Dans le cas des globules rouges, le glutathion réduit empêche l’hémoglobine d’être oxydée en méthémoglobine, laquelle est incapable de transporter l’oxygène.
LFA-3 : leukocyte function-associated antigen ou CD58, dont le ligand est la molécule CD2.
CTLA4 ou CD152 : cytotoxic T-Lymphocyte antigen 4, exprimée à la surface des cellules T activées.
CD80 et CD86, membres de la famille des molécules B27, impliquées dans la coactivation lymphocytaire.
Références
- Schneider CK. Monoclonal antibodies: regulatory challenges. Current Pharmaceutical Biotechnology 2008; 9 :431–8. [Google Scholar]
- Chartrain M, Chu L. Development and production of commercial therapeutic monoclonal antibodies in mammalian cell expression system: an overview of the current upstream technologies. Curr Pharm Biotechnol 2008; 9 : 447–67. [Google Scholar]
- Seamans TC, Fries S, Beck A, et al. Cell cultivation process transfer and scale up in support of the production of early clinical supplies of an anti-IGF-1R antibody (part I). BioProcess Int 2008; 3 : 26. [Google Scholar]
- Beck A, Klinguer-Hamour C, Bussat MC, et al. Peptides as tools and drugs for immunotherapies. J Pept Sci 2007; 13 : 588–602. [Google Scholar]
- Beck A, Wagner-Rousset E, Goetsch L, et al. Therapeutic antibodies: structure assessment by mass spectrometry from screening to clinical batches. Screening Trends Drug Discov 2008; 9 : 18–20. [Google Scholar]
- Reichert JM. Monoclonal antibodies as innovative therapeutics. Curr Pharm Biotechnol 2008; 9 : 423–30. [Google Scholar]
- Beck A, Wagner-Rousset E, Bussat MC, et al. Trends in Glycosylation, Glycoanalysis and Glycoengineering of Therapeutic Antibodies and Fc-Fusion Proteins. Curr Pharm Biotechnol 2008; 9 : 482–501. [Google Scholar]
- Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975; 256 : 495–7. [Google Scholar]
- Morrison SL, Johnson MJ, Herzenberg LA, et al. Chimeric human antibody molecules: mouse antigen-binding domains with human constant region domains. Proc Natl Acad Sci USA 1984; 81 : 6851–5. [Google Scholar]
- Jones PT, Dear PH, Foote J, et al. Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature 1986; 321 : 522–5. [Google Scholar]
- McCafferty J, Griffiths AD, Winter G, et al. Phage antibodies: filamentous phage displaying antibody variable domains. Nature 1990; 348 : 552–4. [Google Scholar]
- Lonberg N, Taylor LD, Harding FA, et al. Antigen-specific human antibodies from mice comprising four distinct genetic modifications. Nature 1994; 368 : 856–9. [Google Scholar]
- Green LL, Hardy MC, Maynard-Currie CE, et al. Antigen-specific human monoclonal antibodies from mice engineered with human Ig heavy and light chain YACs. Nat Genet 1994; 7 : 13–21. [Google Scholar]
- Ruuls SR, Lammerts van Bueren JJ, van de Winkel JG, et al. Novel human antibody therapeutics: the age of the Umabs. Biotechnol J 2008; 3 : 1157–71. [Google Scholar]
- Lonberg N. Fully human antibodies from transgenic mouse and phage display platforms. Curr Opin Immunol 2008; 20 : 450–9. [Google Scholar]
- Peipp M, Dechant M, Valerius T. Effector mechanisms of therapeutic antibodies against ErbB receptors. Curr Opin Immunol 2008; 20 : 436–3. [Google Scholar]
- Jefferis R. Antibody therapeutics: isotype and glycoform selection. Expert Opin Biol Ther 2007; 7 : 1401–13. [Google Scholar]
- Salfeld JG. Isotype selection in antibody engineering. Nat Biotechnol 2007; 25 : 1369–72. [Google Scholar]
- Van der Neut Kolfschoten M., Schuurman J, Losen M, et al. Anti-inflammatory activity of human IgG4 antibodies by dynamic Fab arm exchange. Science 2007; 317 : 1554–7. [Google Scholar]
- Dillon TM, Ricci MS, Vezina C, et al. Structural and functional characterization of disulfide isoforms of the human IgG2 subclass. J Biol Chem 2008; 283 : 16206–15. [Google Scholar]
- Yoo EM, Wims LA, Chan LA, et al. Human IgG2 can form covalent dimers. J Immunol 2003; 170 : 3134–8. [Google Scholar]
- Liu YD, Chen X, Enk JZ, et al. Human IgG2 antibody disulfide rearrangement in vivo. J Biol Chem 2008; 283 : 29266–72. [Google Scholar]
- Presta LG. Molecular engineering and design of therapeutic antibodies. Curr Opin Immunol 2008; 20 : 460–70. [Google Scholar]
- Rother RP, Rollins SA, Mojcik CF, et al. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat Biotechnol 2007; 25 : 1256–64. [Google Scholar]
- Natsume A, In M, Takamura H, et al. Engineered antibodies of IgG1/IgG3 mixed isotype with enhanced cytotoxic activities. Cancer Res 2008; 68 : 3863–72. [Google Scholar]
- Angal S, King DJ, Bodmer MW, et al. A single amino acid substitution abolishes the heterogeneity of chimeric mouse/human (IgG4) antibody. Mol Immunol 1993; 30 : 105–8. [Google Scholar]
- Kai M, Motoki K, Yoshida H, et al. Switching constant domains enhances agonist activities of antibodies to a thrombopoietin receptor. Nat Biotechnol 2008; 26 : 209–11. [Google Scholar]
- Wurch T, Lowe P, Caussanel V, et al. Development of Novel Protein Scaffolds as Alternatives to Whole Antibodies for Imaging and Therapy: Status on Discovery Research and Clinical Validation. Curr Pharm Biotechnol 2008; 9 : 502–9. [Google Scholar]
- Chung CH, Mirakhur B, Chan E, et al. Cetuximab-induced anaphylaxis and IgE specific for galactose-alpha-1,3-galactose. N Engl J Med 2008; 358 : 1109–17. [Google Scholar]
- Wagner-Rousset E, Bednarczyk A, Bussat MC, et al. The way forward, enhanced characterization of therapeutic antibody glycosylation: comparison of three level mass spectrometry-based strategies. J Chromatogr B Analyt Technol Biomed Life Sci 2008; 872 : 23–37. [Google Scholar]
- Satoh M, Iida S, Shitara K. Non-fucosylated therapeutic antibodies as next-generation therapeutic antibodies. Expert Opin Biol Ther 2006; 6 : 1161–73. [Google Scholar]
- Hamilton SR, Gerngross TU. Glycosylation engineering in yeast: the advent of fully humanized yeast. Curr Opin Biotechnol 2007; 18 : 387–92. [Google Scholar]
- Anthony RM, Nimmerjahn F, Ashline DJ, et al. Recapitulation of IVIG anti-inflammatory activity with a recombinant IgG Fc. Science 2008; 320 : 373–6. [Google Scholar]
- Raju TS. Terminal sugars of Fc glycans influence antibody effector functions of IgGs. Curr Opin Immunol 2008; 20 : 471–8. [Google Scholar]
- Labrijn AF, Aalberse RC, Schuurman J. When binding is enough: nonactivating antibody formats. Curr Opin Immunol 2008; 20 : 479–85. [Google Scholar]
- Melmed GY, Targan SR, Yasothan U, et al. Certolizumab pegol. Nat Rev Drug Discov 2008; 7 : 641–2. [Google Scholar]
- Schneider CK, Kalinke U. Toward biosimilar monoclonal antibodies. Nat Biotechnol 2008; 26 : 985–90. [Google Scholar]
- Moon JY, Kim W, Kim JH, et al. A multicenter, randomized, open-label, therapeutic, and exploratory trial to evaluate the tolerability and efficacy of platelet glycoprotein IIb/IIIa receptor blocker (Clotinab) in high-risk patients with percutaneous coronary intervention. Yonsei Med J 2008; 49 : 389–99. [Google Scholar]
- Beck A, Bussat MC, Zorn N, et al. Characterization by liquid chromatography combined with mass spectrometry of monoclonal anti-IGF-1 receptor antibodies produced in CHO and NS0 cells. J Chromatogr B Analyt Technol Biomed Life Sci 2005; 819 : 203–18. [Google Scholar]
- Vlasak J, Ionescu R. Heterogeneity of Monoclonal Antibodies relvealed by charge-sensitive methods. Curr Pharm Biotechnol 2008; 9 : 468–81. [Google Scholar]
- Beck A, Wurch T, Corvaïa N. Editorial: therapeutic antibodies and derivatives: from the bench to the clinic. Curr Pharm Biotechnol 2008; 9 : 421–2. [Google Scholar]
- Swann PG, Tolnay M, Muthukkumar S, et al. Considerations for the development of therapeutic monoclonal antibodies. Curr Opin Immunol 2008; 20 : 493–9. [Google Scholar]
- Kim MS, Lee SH, Song MY, et al. Comparative analyses of complex formation and binding sites between human tumor necrosis factor-alpha and its three antagonists elucidate their different neutralizing mechanisms. J Mol Biol 2007; 374 : 1374–88. [Google Scholar]
- Aggarwal S. What’s fueling the biotech engine-2007. Nat Biotechnol 2008; 26 : 1227–33. [Google Scholar]
- Gires O. EpCAM, une protéine versatile impliquée dans l’oncogenèse. Med Sci (Paris) 2009; 25 : 449–50. [Google Scholar]
- Faye L, Champey Y. Plantes, médicaments et génétique : quelles applications pour demain ? Med Sci (Paris) 2008; 24 : 939–45. [Google Scholar]
- Beck A, Iver H, Reichert JM. European Medecines Agency workshop on biosimilar monoclonal antibodies Meeting Report (London, UK, July 2, 2009). mAbs-Landes Bioscience 2009; 5 : 394–416. (http://www.landesbioscience.com/journals/mabs/article/9630). [Google Scholar]
- Beck A, Cochet O, Wurch T. GlycoFi’s technology to control the glycosylation of recombinant therapeutic proteins. Expert Op Drug Discov 2010 (sous presse). [Google Scholar]
Liste des tableaux
Anticorps monoclonaux et dérivés approuvés pour un usage thérapeutique (FDA, EMEA ou SFDA). * DCI = dénomination commune internationale (www.who.org). Elle est composée par un préfixe unique, suivi d’un infixe représentant la pathologie ciblée (tu = tumeur ; cir = cardiovasculaire ; vi = virus ; lim = immune) et un suffixe lié au type d’AcM [souris = -omab ; chimère = -ximab (souris, rat, primate non humain) ; humanisé = -zumab ; humain = -umab]. La dernière consonne de la syllabe de la cible/ maladie peut être éliminée pour faciliter la prononciation (ex. palivizumab, trastuzumab, rituximab, etc.). Le suffixe des protéines-Fc est -cept ; celui des peptibody -stim. ** Année du 1er enregistrement FDA, EMEA ou SFDA. *** À titre d’exemple, les structures de ces Acm et dérivés sont schématisées sur les Figures 2 et 3. **** Les valeurs des demi-vies et d’immunogénicité sont données à titre indicatif (sources = publications scientifiques, sites web EMEA, FDA et des laboratoires). Elles peuvent varier en fonction des antigènes, des méthodes de dosages, des populations incluses dans les études cliniques et d’autres paramètres (→).
(→) voir G. Paintaud, page 1057 ; L. Manache et al., page 1063 ; P. Stas et I. Lasters, page 1070
Abréviations : CCR = cancer colorectal ; DMLA = dégénérescence maculaire liée à l’âge ; EMEA = European Medicines Agency ; EGFR : epidermal growth factor receptor ; epCAM : epithelial cell adhesion molecule ; FDA = Food and Drug Administration ; HPN = hémoglobinurie paroxystique nocturne ; i.m. = intramusculaire ; i.o. = intraoculaire ; i.p. : intrapéritonéal ; i.v. = intraveineux ; IC = immunoconjugué ; LAM = leucémie aiguë myéloïde ; LLC = leucémie lymphocytaire chronique ; LNH = lymphome non hodgkinien ; PR = polyarthrite rhumatoïde ; PTI = purpura thrombopénique idiopathique ; RIC = radioimmunonoconjugué ; s.c. = sous-cutané ; SA = spondylarthrite ankylosante ; SFDA = China State Food and Drug Administration ; SPAC = syndromes périodiques associés à la cryopyrine ; TNFalpha : tumor necrosis factor alpha ; TPO-R : récepteur de la thrombopoïétine ; VEGF : vascular endothelial growth factor ; NSO, CHO, SP2/20 : lignées cellulaires utilisées pour la production.
Liste des figures
|
Figure 1. hIgG1, chaînes lourde et légère, fragments Fab et Fc. Les région variables sont représentées en bleu clair, les régions constantes en bleu foncé, les CDR en rouge, la région charnière en vert, les ponts disulfure inter-chaînes en brun, les ponts disulfure intra-chaînes en orange et les glycanes en rose. Chaînes lourde (H), légère (L), variable (V), constante (C). |
| Dans le texte | |
|
Figure 2. Exemples d’IgG murines, chimériques, humanisées et humaines (nues et conjuguées). Les exemples illustrés ont été approuvés pour un usage thérapeutique. Les régions variables murines sont représentées en gris clair, les régions constantes murines en gris foncé, les régions variables humaines sont représentées en bleu clair, les régions constantes humaines en bleu foncé, les CDR murins en rouge et humains en bleu foncé, la région charnière murine en vert foncé et humaine en vert clair, les ponts disulfure en orange et les glycanes en rose. |
| Dans le texte | |
|
Figure 3. Exemples de fragments d’Ig approuvés pour un usage thérapeutique (Fab, Fab-pegylé, Fc-protéine et peptide-Fc). Les régions variables humaines sont représentées en bleu clair, les régions constantes en bleu foncé, les CDR en rouge, la région charnière en vert, les résidus PEG en jaune, les ponts disulfure en orange, la partie extracellulaire du récepteur au TNF en bleu et les glycanes en rose. |
| Dans le texte | |
|
Figure 4. Glycoformes des IgG humaines et recombinantes. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.