")
")
| Issue |
Med Sci (Paris)
Volume 42, Number 5, Mai 2026
Agrégation protéique : aux sources des maladies neurodégénératives
|
|
|---|---|---|
| Page(s) | 467 - 475 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/2026065 | |
| Published online | 29 mai 2026 | |
Liens entre diabète de type 2 et maladie de Parkinson
Évidences physiopathologiques et thérapeutiques
Links between type 2 diabetes and Parkinson’s disease: pathophysiological and therapeutic evidence
IGF, Université de Montpellier, CNRS, Inserm, Montpellier, France
# Auteurs correspondants : Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
, Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Le diabète de type 2 et la maladie de Parkinson sont deux maladies associées au vieillissement, chacune étant considérée comme un problème majeur de santé publique dans le monde. Bien que l’une soit identifiée comme un trouble métabolique et l’autre comme une maladie neurodégénérative, des données récentes suggèrent qu’elles pourraient être épidémiologiquement liées. L’accumulation de protéines amyloïdogéniques, la résistance à l’insuline ou encore l’hyperglycémie sont autant de situations pathogéniques qui pourraient expliquer une association épidémiologique et biologique entre ces deux maladies. Cette synthèse vise à décrire les mécanismes d’altération partagés et à explorer les récentes perspectives thérapeutiques communes à ces deux maladies.
Abstract
Type 2 diabetes and Parkinson’s disease are two age-related diseases, each considered a major public health problem worldwide. While one is known as a metabolic disorder and the other as a neurodegenerative disease, recent data suggest that they may be epidemiologically linked. The accumulation of amyloidogenic proteins, insulin resistance, and hyperglycemia are as many pathogenic conditions that could explain an epidemiological and biological association between these two diseases. This synthesis aims to describe the common mechanisms of alteration and explore recent therapeutic perspectives common to both diseases.
Contribution égale
© 2026 médecine/sciences – Inserm
 Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l’utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l’utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.

Vignette (© Wikimedia Commons - Boku wa Kage).
Physiopathologie
Le diabète de type 2 est caractérisé par une hyperglycémie chronique qui s’explique par une diminution de la sensibilité à l’insuline au niveau des tissus cibles (foie, muscle, tissu adipeux), associée à un défaut de sécrétion d’insuline par les cellules β pancréatiques des îlots de Langerhans1. Dans le diabète de type 2 chez l’être humain, l’îlot est caractérisé par des dépôts amyloïdes dérivant du polypeptide amyloïde de l’îlot (islet amyloid polypeptide [IAPP], composé de 37 acides aminés) [1], une protéine coexprimée et cosécrétée avec l’insuline exclusivement par les cellules β [1]. Bien que ses rôles physiologiques ne soient pas complètement compris, l’IAPP contribuerait au ralentissement de la vidange gastrique et au contrôle de la satiété, mais également à l’inhibition de la sécrétion d’insuline grâce à un effet paracrine direct sur les cellules β. Cependant, les situations à l’origine d’une augmentation compensatoire de la production d’insuline - comme la résistance à l’insuline et l’hyperglycémie - favorisent une augmentation de la production d’IAPP [1]. Cette augmentation des niveaux d’expression de l’IAPP au-delà d’un certain seuil (on parle alors de « protéotoxicité ») altère la fonction et la survie des cellules β, conduisant au diabète de type 2 [1].
La maladie de Parkinson est, quant à elle, une maladie neurodégénérative très fréquente qui débute cinq à dix ans avant l’apparition des premiers signes cliniques. Ses principaux symptômes sont les tremblements au repos, une bradykinésie2, une instabilité posturale et une rigidité, accompagnés par un certain nombre de symptômes non moteurs, tels que des troubles du sommeil, un dysfonctionnement olfactif, la dépression ou encore la constipation [2]. Elle est principalement caractérisée par une dégénérescence progressive des neurones dopaminergiques de la substance noire pars compacta et de leurs prolongements dans le striatum3. Ses lésions caractéristiques, les corps et neurites de Lewy4, contiennent une protéine amyloïdogénique, l’alphasynucléine (αS, composée de 140 acides aminés), qui, en conditions physiologiques, semble jouer un rôle dans la plasticité synaptique (trafic/fusion vésiculaire, libération de neurotransmetteurs, intégrité des synapses), mais qui, en conditions physiopathologiques, est capable de s’agréger et de s’accumuler sous forme oligomérique et fibrillaire. Une modification post-traductionnelle majeure de l’αS est l’abondante phosphorylation d’une sérine en position 129, un marqueur des « synucléinopathies ».
Association épidémiologique entre le diabète de type 2 et la maladie de Parkinson
Plusieurs études prospectives de grandes cohortes rapportent que le diabète de type 2 et la maladie de Parkinson sont associés sur le plan épidémiologique [3–5], avec un risque accru de développer la maladie de Parkinson chez les patients atteints de diabète de type 2 [3, 4]. Une méta-analyse regroupant plus de 30 études estime que le risque de développer la maladie de Parkinson chez les patients diabétiques est augmenté en moyenne de 27 % à 40 % [6], avec notamment une évolution plus rapide et un phénotype plus sévère de la maladie de Parkinson [7]. Bien que la prévalence du diabète de type 2 ne semble pas plus élevée chez les personnes atteintes de la maladie de Parkinson, certaines études rapportent des altérations de la tolérance au glucose, une résistance à l’insuline ou des hyperglycémies postprandiales fréquentes chez ces personnes [8], suggérant, là encore, un lien entre ces deux maladies (Figure 1).
 |
Figure 1 Liens entre diabète de type 2 et maladie de Parkinson. |
Mécanismes d’altération communs à la maladie de Parkinson et au diabète de type 2
Au-delà du fait que le diabète de type 2 (hyperglycémie) semble accélérer la progression de la maladie de Parkinson, certains processus initiateurs communs (agrégation de protéines amyloïdogéniques, résistance à l’insuline, etc.) pourraient également être impliqués dans cette association (Figures 1 et 2). Ainsi, une prédisposition génétique commune et/ou l’existence de mécanismes pathogéniques communs (Figure 2) pourraient expliquer le développement concomitant de ces deux maladies. Ces mécanismes seront décrits dans les paragraphes suivants.
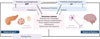 |
Figure 2 Évidences physiopathologiques et mécanistiques de l’association entre le diabète de type 2 et la maladie de Parkinson. L’agrégation des protéines amyloïdogéniques, IAPP et αS, dans leurs types cellulaires respectifs (et leur co-agrégation/cross-seeding dans les cellules β et les neurones), la résistance à l’insuline (centrale et/ou périphérique) ainsi que l’hyperglycémie conduisent à des mécanismes cellulaires communs d’altération : perturbation de l’autophagie, stress du réticulum endoplasmique, stress oxydant/mitochondrial et inflammation lesquels aboutissent à la dysfonction et/ou à la mort cellulaire. IAPP: islet amyloid polypeptide; αS: alpha-synucléine. |
Ces deux maladies résultent in fine de la perte fonctionnelle et progressive d’une population cellulaire spécifique – les cellules β pancréatiques pour le diabète de type 2 et les neurones dopaminergiques de la substance noire pars compacta pour la maladie de Parkinson – bien avant l’apparition des symptômes cliniques. Dans le diabète de type 2, une perte moyenne de 40 % de la masse des cellules β pancréatiques est observée (24 % pour une durée du diabète inférieure à 5 ans ; 54 % au-delà de 15 ans) [9] ; dans la maladie de Parkinson, la perte neuronale peut atteindre 50 à 70 % avant que les premiers défauts moteurs n’apparaissent.
Les protéines amyloïdogéniques
Le diabète de type 2 et la maladie de Parkinson appartiennent au groupe des maladies à « mauvais repliement protéique », dont l’agrégation de protéines amyloïdogéniques est l’une des principales caractéristiques. Ce type d’altération est typiquement observé dans des cellules à longue durée de vie, comme les neurones et les cellules β pancréatiques, entraînant une dysfonction et une mort cellulaire.
L’IAPP humain (contrairement à la forme murine) et l’αS, comme d’autres protéines amyloïdogéniques, sont des protéines intrinsèquement désordonnées5, capables d’adopter diverses conformations qui facilitent les interactions moléculaires [10]. Cette propriété favorise leur propension à acquérir des conformations anormales, conduisant à la formation d’oligomères toxiques et de fibres amyloïdes. D’autre part, les formes mal repliées de ces protéines peuvent propager leur conformation pathologique non seulement à leurs monomères natifs, mais également à d’autres protéines amyloïdogéniques par un phénomène de cross-seeding, alimentant ainsi un cycle d’agrégation similaire à celui observé pour les protéines prions [11, 12].
Les mécanismes cellulaires d’altération engagés spécifiquement par l’IAPP humain et l’αS sont brièvement décrits dans les parties suivantes.
Co-agrégation et cross-seeding de l’αS et de l’IAPP humain
De manière intéressante, l’αS, principalement produite par les neurones, est aussi exprimée dans les cellules β et pourrait jouer un rôle délétère sur la sécrétion d’insuline et la survie de ces cellules [13, 14], même si un rôle physiologique dans les cellules endocrines du pancréas a également été décrit [14, 15]. Dans un contexte pathologique, l’αS a été détectée sous forme agrégée et phosphorylée sur la sérine en position 129 dans les îlots pancréatiques de singes cynomolgus développant un diabète de type 2 spontané [16], ainsi que dans les cellules β de sujets humains atteints de diabète de type 2, qu’ils soient ou non atteints de la maladie de Parkinson [14, 17–19]. Des études montrent notamment une co-localisation [16–20], une interaction moléculaire directe et une co-agrégation des protéines αS et IAPP dans les îlots [17, 18, 20]. L’αS sous forme fibrillaire a aussi la capacité d’induire l’agrégation d’IAPP in vitro grâce à un mécanisme de crossseeding [19–22], et l’injection intraveineuse d’αS monomérique chez des souris transgéniques surexprimant l’IAPP humain/amyloïdogénique exacerbe l’agrégation de l’IAPP in vivo [19].
Contrairement à l’αS, qui est exprimée aussi bien dans le cerveau que dans les cellules β, l’IAPP est synthétisé uniquement par les cellules β [1]. L’IAPP peut cependant passer la barrière hématoencéphalique [23] et a notamment été détecté dans les corps de Lewy des patients atteints de la maladie de Parkinson où il interagit avec l’αS [21]. Cette étude révèle également que l’injection intraveineuse d’IAPP sous forme fibrillaire chez des souris modèles de synucléinopathie exacerbe l’agrégation de l’αS et favorise ainsi l’apparition de troubles moteurs caractéristiques de la maladie de Parkinson [21]. Ces processus de co-agrégation et cross-seeding engagés par l’IAPP pour accélérer l’agrégation de l’αS ont également été mis en évidence in vitro [22], illustrant encore une fois ce crosstalk amyloïdogénique.
L’ensemble de ces études souligne donc le fait que l’IAPP et l’αS interagissent étroitement en co-agrégeant à la fois dans le cerveau et dans le pancréas, ainsi qu’en propageant leur toxicité de façon réciproque par des mécanismes de cross-seeding. Ces phénomènes pourraient, entre autres, expliquer certains liens entre la maladie de Parkinson et le diabète de type 2.
Altération de l’autophagie
Les cellules β et les neurones supportent une charge importante en matière de synthèse et de conformation des protéines. La dégradation des protéines mal repliées est primordiale pour ces cellules. L’autophagie est un processus cellulaire essentiel permettant, entre autres, le recyclage des protéines mal conformées, des organites endommagés et des protéines amyloïdogéniques, comme l’IAPP et l’αS. Au cours de ce processus, le matériel séquestré au sein d’organelles appelés autophagosomes est dégradé après la fusion des autophagosomes avec les lysosomes. Un défaut de formation des autophagosomes et/ou de la dégradation lysosomale résulte en une autophagie déficiente. L’altération de cette voie entraîne d’ailleurs une accumulation de l’IAPP dans les cellules β [24] et de l’αS dans les neurones [25]. Réciproquement, l’IAPP et l’αS contribuent, elles-mêmes, à l’altération de l’autophagie dans les cellules β [14, 26] et dans les neurones [27], engageant ainsi un cercle vicieux qui accroît leur accumulation. L’observation de marqueurs d’altération de l’autophagie à la fois dans les cellules β de sujets atteints de diabète de type 2 [28] et dans les neurones dopaminergiques de sujets atteints de la maladie de Parkinson [25] renforce la pertinence de ces mécanismes chez l’être humain ainsi que les liens biologiques entre ces maladies.
Stress du réticulum endoplasmique
Le stress du réticulum endoplasmique est une altération bien décrite dans le diabète de type 2 et la maladie de Parkinson [29, 30]. En effet, l’accumulation de protéines mal repliées dans le lumen du réticulum endoplasmique des cellules β ou des neurones dopaminergiques engage une cascade d’évènements qui conduit in fine à la mort cellulaire par apoptose. Par ailleurs, les oligomères toxiques (d’IAPP ou d’αS) ont la capacité d’endommager la membrane des organites intracellulaires, ce qui compromet l’intégrité du réticulum endoplasmique et contribue ainsi à ce stress cellulaire [1, 30]. Le stress du réticulum endoplasmique est également associé à l’altération d’une autre voie de dégradation protéique, cruciale pour les cellules β et les neurones : le système ubiquitine-protéasome [1, 30].
Dysfonction mitochondriale et stress oxydant
La dysfonction mitochondriale et le stress oxydant, définis comme une production et une accumulation excessive d’espèces réactives de l’oxygène, sont également des médiateurs de l’altération des cellules β induite par l’IAPP [29], ainsi que de celle des neurones dopaminergiques induite par l’αS [31]. Réciproquement, le stress oxydant favorise l’oxydation et la modification de l’αS, lesquelles perturbent son repliement normal et favorisent la formation d’oligomères toxiques [32]. De manière similaire, le stress oxydatif semble également contribuer à l’agrégation de l’IAPP [33].
Inflammation
La toxicité des protéines amyloïdogéniques est associée à l’initiation de processus inflammatoires, caractéristiques de la pathologie de l’îlot dans le diabète de type 2 [29], mais aussi des altérations neuronales dans la maladie de Parkinson [34]. L’activation d’un complexe protéique appelé inflammasome [35] (→) par l’αS ou par l’IAPP déclenche l’activation de la caspase-1 et la maturation/production de cytokines pro-inflammatoires, respectivement par les cellules microgliales [34] et les macrophages infiltrés des îlots [29]. Ces cytokines altèrent l’intégrité des cellules β [29] et conduisent à la dégénérescence des neurones dopaminergiques [34].
(→) Voir m/s n° 1, 2018, page 47
L’insulinorésistance
L’insulinorésistance, un facteur de risque majeur pour le diabète de type 2, correspond à une diminution de l’action de l’insuline sur ses tissus cibles (foie, muscle, tissu adipeux). Pour compenser cette situation, les cellules β ont la capacité de s’adapter en augmentant leur sécrétion d’insuline. Cependant, cette augmentation de sécrétion d’insuline est également associée à une synthèse accrue d’IAPP. Au-delà d’un certain seuil, lorsque la capacité du réticulum endoplasmique en termes de synthèse/de repliement/et de transport des protéines est saturée, l’IAPP s’accumulerait sous forme d’oligomères toxiques, induisant une « protéotoxicité » qui compromet la fonction sécrétoire et la survie des cellules β [1]. Le diabète de type 2 et l’hyperglycémie s’installent quand les cellules β ne peuvent plus faire face aux besoins insuliniques de l’organisme. Cette situation est aggravée par la diminution, au cours du temps, du nombre de cellules β [29].
Plusieurs études relatent une association entre l’insulinorésistance périphérique et l’augmentation de la prévalence de la maladie de Parkinson, mais les mécanismes impliqués ne sont pas réellement connus. Par ailleurs, étant donné la présence de récepteurs à l’insuline au niveau du système nerveux central, potentiellement à l’origine d’effets neurotrophiques et neuroprotecteurs, une insulinorésistance centrale pourrait également être impliquée dans la pathogenèse de la maladie de Parkinson [36] (→).
(→) Voir m/s n° 4, 2009, page 337
Une diminution du nombre de récepteurs à l’insuline a d’ailleurs été spécifiquement observée dans la substance noire de sujets atteints de la maladie de Parkinson, et ce, en lien avec une diminution de la tyrosine hydroxylase (enzyme impliquée dans la synthèse de dopamine) [8]. L’invalidation spécifique du récepteur à l’insuline dans le cerveau de souris entraîne, entre autres, une altération de la fonction dopaminergique, une augmentation de la prise alimentaire et une insulinorésistance périphérique [37]. Ces observations supportent l’implication d’une insulinorésistance centrale dans le développement de la maladie de Parkinson. Bien qu’ils ne soient pas totalement compris, les mécanismes centraux induits par l’altération de la signalisation insulinique impliqueraient la neuroinflammation, le stress oxydant et une augmentation des dépôts d’αS contribuant à une mort neuronale accélérée. En effet, des souris modèles de synucléinopathie rendues obèses et prédiabétiques par un régime riche en graisses (modèle d’insulinorésistance) présentent une apparition plus rapide des troubles moteurs, accompagnée d’une inflammation, de l’accumulation d’αS phosphorylée sur la sérine en position 129, et de dépôts amyloïdes dans le tronc cérébral, ainsi qu’une signalisation centrale insulinique altérée [38].
Ainsi, l’insulinorésistance, déjà considérée comme un facteur de risque pour le diabète de type 2, pourrait également être un acteur dans le développement et la progression de la maladie de Parkinson.
L’hyperglycémie
Une fois le diagnostic du diabète établi, les taux élevés et persistants de glucose détectés chez les personnes atteintes de diabète de type 2 contribuent aux complications diabétiques. Cette hyperglycémie peut également exacerber la perte de cellules β fonctionnelles, un concept appelé « glucotoxicité » [29]. Les mécanismes impliqués regroupent le stress du réticulum endoplasmique, la dysfonction mitochondriale, l’altération des voies de dégradation protéique et l’inflammation [29]. Au-delà de ses effets délétères sur les cellules β, l’hyperglycémie chronique aurait un impact direct sur le système nerveux central. En effet, plusieurs modèles expérimentaux ont montré que la dérégulation glycémique potentialise la toxicité de l’αS et accélère la progression de la maladie de Parkinson. À titre d’exemple, l’hyperglycémie persistante induite par la destruction ciblée des cellules β chez des souris surexprimant l’αS aggrave la perte des neurones dopaminergiques, favorise l’agrégation protéique, et contribue à l’exacerbation des déficits moteurs [39]. Ainsi, au-delà de l’insulinorésistance, l’hyperglycémie seule aurait des effets majeurs sur la progression de la maladie de Parkinson.
D’une manière générale, l’hyperglycémie est à l’origine de la glycation des protéines (modification spontanée non enzymatique) qui conduit à la formation d’AGE (advanced glycation end-products) dont le potentiel néfaste participe à la pathogenèse du diabète de type 2 et à ses complications. Les AGE contribuent notamment à l’insulinorésistance ainsi qu’à l’altération des cellules β [40]. Une étude in vitro a également suggéré que la glycation de l’IAPP favoriserait son potentiel amyloïdogénique [41]. De manière intéressante, les intermédiaires préamyloïdes de l’IAPP peuvent aussi interagir avec les récepteurs aux AGE (RAGE : receptor for advanced glycation end-products) pour exercer leurs effets toxiques sur les cellules β [42]. Dans les neurones, la glycation de la forme monomérique de l’αS accélère également son agrégation, la formation de fibres et d’oligomères toxiques résistants à la dégradation protéique [43]. En conséquence, les souris modèles de la maladie de Parkinson qui sont exposées à une glycation accrue présentent une perte neuronale et des déficits moteurs accentués [43]. La pertinence de ce mécanisme chez l’être humain est confortée par la détection d’αS glyquée chez des patients atteints de la maladie de Parkinson [43], démontrant là encore un lien physiopathologique entre les différentes phases évolutives du diabète de type 2 et la maladie de Parkinson.
Repositionnement thérapeutique de certains antidiabétiques pour la maladie de Parkinson ?
Étant donné l’association entre les deux maladies, avec un risque accru de développer la maladie de Parkinson, une évolution plus rapide de celle-ci chez des patients atteints de diabète de type 2, ainsi que l’existence de mécanismes d’altération communs, la question se pose de savoir si des antidiabétiques peuvent ralentir la progression de la maladie de Parkinson. À ce jour, les traitements disponibles pour la maladie de Parkinson visent à soulager les symptômes (moteurs ou non moteurs). Pour les symptômes moteurs, les thérapies reposent principalement sur les agonistes de la dopamine qui miment les effets du neurotransmetteur au niveau des neurones, ou des inhibiteurs de la monoamine oxydase B qui permettent de réduire la dégradation de la dopamine [44]. Cependant, il n’existe actuellement aucune thérapie efficace pour ralentir la progression de la maladie de Parkinson, et c’est dans ce contexte qu’un repositionnement des antidiabétiques est à l’étude. Seules les principales thérapies du diabète de type 2 présentant un intérêt dans le contexte de la maladie de Parkinson seront citées ci-dessous.
Les thérapies actuelles du diabète de type 2
Thérapies ciblant le foie et les reins
Suivant les recommandations actuelles de la Haute Autorité de Santé [45], au-delà du changement de certaines habitudes de vie (alimentation, activité physique, etc.), la metformine constitue le traitement de première intention pour les patients atteints de diabète de type 2, afin de réduire la résistance périphérique à l’insuline, en agissant principalement sur le foie. D’autres thérapies peuvent venir compléter le traitement par metformine, comme les inhibiteurs du co-transporteur sodium/glucose 2 (iSGLT2) ou gliflozines, qui permettent d’éliminer le surplus de glucose directement dans les urines.
Thérapies ciblant le pancréas et les organes insulino-sensibles
Une stratégie thérapeutique consiste à stimuler la sécrétion d’insuline par les cellules β pancréatiques. À cet égard, les agonistes du récepteur au GLP-1 (glucagon-like peptide-1, GLP-1RA) sont recommandés pour augmenter la sécrétion d’insuline par les cellules β sans risque d’hypoglycémies (Figure 3), notamment lorsque la réduction du poids est également recherchée. Il est important de noter que les agonistes d’ancienne génération à courte durée d’action (1 injection par jour) sont actuellement beaucoup moins utilisés (voire arrêtés) et ont laissé la place aux GLP-1RA de dernière génération (action prolongée, 1 injection par semaine) [45, 46] (→).
(→) Voir m/s n° 11, 2024, page 837
 |
Figure 3 Repositionnement thérapeutique des antidiabétiques GLP-1RA pour la maladie de Parkinson : évidences précliniques et cliniques. Les études précliniques réalisées chez les rongeurs montrent que l’utilisation des GLP-1RA entraîne une augmentation de la sécrétion d’insuline et une diminution de l’apoptose des cellules β pancréatiques et des neurones dopaminergiques par l’intermédiaire de certains mécanismes communs. Les GLP-1RA utilisés pour le traitement du diabète de type 2 favorisent une diminution de la résistance à l’insuline et de l’hyperglycémie. En ce qui concerne la maladie de Parkinson, les essais cliniques impliquant les GLP-1RA montrent un ralentissement de l’apparition et de la progression des troubles moteurs. |
Les GLP-1RA pourraient également protéger les cellules β de la mort par apoptose induite par une gluco- et lipotoxicité (concentrations élevées de glucose et de palmitate) ou par des cytokines [29] (Figure 3). Plusieurs mécanismes semblent être impliqués dans cette protection (Figure 3). En effet, les GLP-1RA permettent notamment l’activation du facteur de transcription CREB (cAMP response element-binding protein 1) [47], qui induit la transcription de gènes de survie pour la cellule β [29]. Les GLP-1RA permettraient également de rétablir le flux de l’autophagie dans les cellules β soumises à des conditions diabétogènes [29], et de réduire le stress du réticulum endoplasmique [29]. Plus récemment, des doubles agonistes des récepteurs au GLP-1 et au GIP (glucose-dependent insulinotropic polypeptide) ont également reçu l’autorisation pour le traitement du diabète de type 2 et de l’obésité. En effet, le GIP ayant des effets complémentaires à ceux du GLP-1 sur le tissu adipeux et au niveau du système nerveux central [48], son utilisation a été reconsidérée pour une association thérapeutique avec le GLP-1 combinant ainsi les effets des deux peptides en une seule molécule, le tirzepatide (TZP), qui est, à ce jour, l’unique double agoniste utilisé pour le traitement du diabète de type 2 et de l’obésité [46]. Le TZP améliore davantage la sensibilité à l’insuline et la perte de poids, en comparaison avec les GLP-1RA utilisés comme mono-agonistes.
Les effets des antidiabétiques dans la maladie de Parkinson
Évidences précliniques
Puisque le récepteur au GLP-1 (GLP-1R) est retrouvé au niveau du système nerveux central chez l’être humain et les rongeurs, plusieurs études précliniques ont étudié l’effet neuroprotecteur de différents GLP-1RA dans le contexte de la maladie de Parkinson chez les rongeurs (Figure 3). Concernant les GLP-1RA à courte durée d’action, l’exendine-4 (Ex-4 ; le premier GLP-1RA mis sur le marché) a montré des effets bénéfiques dans plusieurs modèles de rongeurs précliniques de la maladie de Parkinson (induits pharmacologiquement par les neurotoxines MPTP [tétrahydropyridine : 1méthyl-4phényl -1,2,3,6-tétrahydro pyridine] et 6-OHDA [6-hydroxydopamine]), en freinant la dégénérescence des neurones dopaminergiques de la substance noire pars compacta, en préservant les niveaux de dopamine et en améliorant les fonctions motrices [49–51]. Le Lixisénatide (autre analogue de l’Ex-4), quant à lui, réduit la phosphorylation d’αS sur la sérine en position 129, son agrégation et sa propagation [52]. Pour les GLP-1RA à durée d’action prolongée, le NLY01 (analogue de l’Ex-4 ayant subi une PEGylation lui permettant de mieux traverser la barrière hématoencéphalique) a également montré des effets bénéfiques en réduisant l’accumulation d’αS dans les souris modèles de synucléinopathie (souris génétiquement modifiées avec une mutation A53T [adénine en position 53 mutée pour une thymine]) [53]. Enfin, le Sémaglutide (GLP-1RA de dernière génération) a induit des effets bénéfiques supérieurs au Liraglutide (courte durée d’action) en réduisant davantage les défauts moteurs, l’accumulation d’αS et l’inflammation du système nerveux central chez des souris MPTP [54]. Il est important de souligner que même si certains GLP-1RA ne semblent pas traverser la barrière hémato-encéphalique (tel que le Sémaglutide) [55], ils agissent tous au niveau du système nerveux central [56] et semblent ralentir la progression de la maladie de Parkinson. Néanmoins les mécanismes impliqués restent encore à déterminer.
Plusieurs études précliniques se sont également intéressées aux effets des doubles agonistes GLP-1/GIP non commercialisés (DA5-CH et DA3-CH), qu’ils ont comparés aux GLP-1RA dans différents modèles de la maladie de Parkinson (6-OHDA chez le rat, MPTP et mutation A53T chez la souris). Ils ont permis d’améliorer les défauts moteurs, de réduire l’accumulation d’αS, de freiner la perte des neurones dopaminergiques dans la substance noire pars compacta, de réduire l’inflammation, de diminuer l’apoptose, d’améliorer l’autophagie de manière plus importante qu’un GLP-1RA seul comme le Liraglutide [57–59] ou le Sémaglutide [60]. Plus récemment, le double agoniste TZP a également montré des résultats positifs significatifs dans l’amélioration des signes moteurs, la diminution de la dégénérescence des neurones dopaminergiques de la substance noire pars compacta, la réduction de l’agrégation d’αS, de l’inflammation et du stress oxydatif ou de l’intégrité mitochondriale au niveau du système nerveux central dans des modèles de la maladie de Parkinson induits par la roténone [61] ou MPTP [62].
Évidences cliniques
Plusieurs critères d’évaluation doivent être pris en compte lorsqu’il s’agit d’étudier les effets, chez les patients, des antidiabétiques dans un contexte de maladie de Parkinson. Il faut étudier l’évolution des signes moteurs et non moteurs, l’apparition d’effets indésirables qui peuvent être liés aux médicaments, ainsi que l’amélioration de la qualité de vie du patient. Quelques études ont mesuré sur plusieurs années l’incidence ou l’apparition de la maladie de Parkinson chez des patients diagnostiqués pour le diabète de type 2, et ont rapporté que celle-ci varie considérablement en fonction du traitement antidiabétique reçu [63, 64]. Les médicaments antidiabétiques ciblant l’insulinorésistance (metformine) [65] ou faisant baisser la glycémie en éliminant le sucre dans les urines (iSGLT2) [66] pourraient réduire l’incidence de la maladie de Parkinson chez les personnes atteintes de diabète de type 2, avec un effet plus marqué pour les iSGLT2 [65]. Néanmoins d’autres études cliniques sont nécessaires pour pouvoir confirmer ces effets. En revanche l’utilisation de GLP-1RA est associée à un taux plus faible d’incidence de la maladie de Parkinson par rapport à l’utilisation de tout autre antidiabétique [63, 64, 67].
Depuis deux ans, un espoir de voir ralentir la progression de la maladie de Parkinson avec les antidiabétiques GLP-1RA, est donc apparu. Pour l’instant, les études réalisées rapportent des effets avec les agonistes d’ancienne génération à courte durée d’action (1 injection par jour). L’Exénatide (version commerciale de l’Ex-4, à courte durée d’action) a été le premier agoniste du GLP-1R à avoir été évalué en clinique chez des patients atteints de la maladie de Parkinson [68–70]. D’autres études utilisant des analogues similaires comme le NLY01 [71] ou le Lixisénatide [72] ont également été conduites. Le Lixisénatide administré pendant 12 mois a entraîné une progression moindre des défauts moteurs chez des personnes atteintes de la maladie de Parkinson (diagnostiquées depuis moins de trois mois) [72]. Les traitements à l’Exénatide ont donné des résultats plus contrastés, avec des effets positifs [68, 69] ou aucun effet [70]. Néanmoins, il semblerait que dans cette dernière étude, les patients ne présentaient pas d’insulinorésistance. De façon générale, les effets indésirables rencontrés sont d’ordre gastro-intestinal, comme attendu avec cette classe de médicaments (ralentissement de la vidange gastrique et diminution de l’appétit). Toutes ces études semblent montrer un effet bénéfique des GLP-1RA sur le ralentissement de la progression de la maladie de Parkinson (Figure 3), mais des essais cliniques plus longs et à plus grande échelle sont maintenant nécessaires pour confirmer ces résultats. Par ailleurs, il n’existe pas de données concernant les GLP-1RA de dernière génération (action prolongée, 1 injection par semaine) chez des patients atteints de diabète de type 2, même si une réduction du risque de développer la maladie de Parkinson a été rapportée chez des patients obèses traités au Sémaglutide [73].
Étant donné le récent développement des doubles agonistes GLP-1/GIP, il n’existe pas encore d’étude clinique évaluant les effets de ces traitements sur des patients atteints de la maladie de Parkinson. Cependant, les études précliniques ayant rapporté des résultats prometteurs, il ne fait aucun doute que l’impact de doubles agonistes sera prochainement évalué dans le contexte clinique de la maladie de Parkinson.
Conclusion
L’association épidémiologique rapportée entre diabète de type 2 et maladie de Parkinson semble s’expliquer par l’existence de plusieurs situations pathogéniques communes (agrégation de protéines amyloïdogéniques, insulinorésistance) ou aggravantes (hyperglycémie). Sur le plan mécanistique, là encore, les principaux acteurs de ces maladies (cellule β pancréatiques et neurones dopaminergiques) sont le théâtre d’altérations cellulaires communes au diabète de type 2 et à la maladie de Parkinson. Les causes de ces maladies liées à l’âge étant multifactorielles, l’impact des facteurs environnementaux jouerait également un rôle important dans leur prévalence. Bien que cet aspect n’ait pas été abordé dans cette synthèse, l’alimentation, la sédentarité, l’exposition chronique aux polluants environnementaux pourraient contribuer aux altérations communes qui sont décrites, et pourraient fournir des éléments d’explication supplémentaires à l’association entre ces deux maladies. Par ailleurs, le diabète de type 2 touche une population de plus en plus jeune, ce qui, dans un contexte d’augmentation du risque de développer la maladie de Parkinson, peut s’avérer être un réel problème de santé publique. D’un point de vue thérapeutique, le diabète de type 2 et la maladie de Parkinson semblent, là encore, intrinsèquement liés, puisqu’un repositionnement des antidiabétiques actuels offre de nouveaux espoirs pour ralentir la progression de la maladie de Parkinson. Il reste cependant à mieux comprendre si les effets bénéfiques de ces nouvelles thérapies repositionnées pour la maladie de Parkinson s’appuient sur une amélioration des conditions métaboliques et/ou sur des mécanismes neuroprotecteurs directs. Le développement de modèles précliniques récapitulant à la fois le diabète de type 2 et la maladie de Parkinson est maintenant nécessaire pour mieux évaluer au niveau moléculaire les mécanismes impliqués dans l’exacerbation réciproque de ces deux maladies, mais également pour déterminer comment les GLP-1RA exercent leurs effets neuroprotecteurs dans un contexte de diabète de type 2.
Liens d’intérêt
Les auteurs déclarent ne pas avoir de liens d’intérêt.
Remerciements
Nous remercions Thierry Baron (ANSES, Lyon) pour la relecture critique du manuscrit, ainsi que les agences de financements : l’Agence nationale de sécurité sanitaire de l’alimentation, de l’environnement et du travail (ANSES, DEEP EST-21-217 attribuée à SC), l’Agence nationale de la recherche (ANR PESTILOID attribuée à SC ; ANR DiaPARKprotect attribuée à MAR), la Société francophone du diabète (SFD, attribuées à SC et à MAR) et la fondation France Parkinson (Bourse doctorale attribuée à AA).
Références
- Costes S, Langen R, Gurlo T, et al. beta-Cell failure in type 2 diabetes: a case of asking too much of too few? Diabetes 2013; 62 : 327–35. [Google Scholar]
- Dickson DW, Braak H, Duda JE, et al. Neuropathological assessment of Parkinson’s disease: refining the diagnostic criteria. Lancet Neurol 2009; 8 : 1150–7. [Google Scholar]
- Cheong JLY, de Pablo-Fernandez E, Foltynie T, Noyce AJ. The association between type 2 diabetes mellitus and Parkinson’s disease. J Parkinsons Dis 2020; 10 : 775–89. [Google Scholar]
- De Pablo-Fernandez E, Goldacre R, Pakpoor J, et al. Association between diabetes and subsequent Parkinson disease: A record-linkage cohort study. Neurology 2018; 91 : e139–e42. [Google Scholar]
- Hu G, Jousilahti P, Bidel S, et al. Type 2 diabetes and the risk of Parkinson’s disease. Diabetes Care 2007; 30 : 842–7. [Google Scholar]
- Yue X, Li H, Yan H, et al. Risk of Parkinson disease in diabetes mellitus: an updated meta-analysis of population-based cohort studies. Medicine (Baltimore) 2016; 95 : e3549. [Google Scholar]
- Chohan H, Senkevich K, Patel RK, et al. Type 2 diabetes as a determinant of parkinson’s disease risk and progression. Mov Disord 2021; 36 : 1420–9. [Google Scholar]
- Cullinane PW, de Pablo Fernandez E, Konig A, et al. Type 2 diabetes and Parkinson’s disease: a focused review of current concepts. Mov Disord 2023; 38 : 162–77. [Google Scholar]
- Rahier J, Guiot Y, Goebbels RM, et al. Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes Metab 2008; 10 (suppl 4) : 32–42. [Google Scholar]
- Nguyen PH, Ramamoorthy A, Sahoo BR, et al. Amyloid oligomers: a joint experimental/computational perspective on alzheimer’s disease, Parkinson’s disease, type ii diabetes, and amyotrophic lateral sclerosis. Chem Rev 2021; 121 : 2545–647. [Google Scholar]
- Mougenot AL, Nicot S, Bencsik A, et al. Prion-like acceleration of a synucleinopathy in a transgenic mouse model. Neurobiol Aging 2012; 33 : 2225–8. [Google Scholar]
- Mukherjee A, Morales-Scheihing D, Salvadores N, et al. Induction of IAPP amyloid deposition and associated diabetic abnormalities by a prion-like mechanism. J Exp Med 2017; 214 : 2591–610. [Google Scholar]
- Geng X, Lou H, Wang J, et al. alpha-Synuclein binds the K(ATP) channel at insulin-secretory granules and inhibits insulin secretion. Am J Physiol Endocrinol Metab 2011; 300 : E276–86. [Google Scholar]
- Steneberg P, Bernardo L, Edfalk S, et al. The type 2 diabetes-associated gene ide is required for insulin secretion and suppression of alpha-synuclein levels in beta-cells. Diabetes 2013; 62 : 2004–14. [Google Scholar]
- Wijesekara N, Ahrens R, Wu L, et al. Alpha-synuclein regulates peripheral insulin secretion and glucose transport. Front Aging Neurosci 2021; 13 : 665348. [Google Scholar]
- Sun Y, Guo C, Yuan L, et al. Cynomolgus monkeys with spontaneous type-2-diabetes-mellitus-like pathology develop alpha-synuclein alterations reminiscent of prodromal Parkinson’s disease and related diseases. Front Neurosci 2020; 14 : 63. [Google Scholar]
- Martinez-Valbuena I, Amat-Villegas I, Valenti-Azcarate R, et al. Interaction of amyloidogenic proteins in pancreatic beta cells from subjects with synucleinopathies. Acta Neuropathol 2018; 135 : 877–86. [Google Scholar]
- Martinez-Valbuena I, Valenti-Azcarate R, Amat-Villegas I, et al. Mixed pathologies in pancreatic beta cells from subjects with neurodegenerative diseases and their interaction with prion protein. Acta Neuropathol Commun 2021; 9 : 64. [Google Scholar]
- Mucibabic M, Steneberg P, Lidh E, et al. alpha-Synuclein promotes IAPP fibril formation in vitro and beta-cell amyloid formation in vivo in mice. Sci Rep 2020; 10 : 20438. [Google Scholar]
- Wang Y, Bergstrom J, Ingelsson M, Westermark GT. Studies on alpha-synuclein and islet amyloid polypeptide interaction. Front Mol Biosci 2023; 10 : 1080112. [Google Scholar]
- Meng L, Li Y, Liu C, et al. Islet amyloid polypeptide triggers alpha-synuclein pathology in Parkinson’s disease. Prog Neurobiol 2023; 226 : 102462. [Google Scholar]
- Horvath I, Wittung-Stafshede P. Cross-talk between amyloidogenic proteins in type-2 diabetes and Parkinson’s disease. Proc Natl Acad Sci USA 2016; 113 : 12473–7. [Google Scholar]
- Banks WA, Kastin AJ. Differential permeability of the blood-brain barrier to two pancreatic peptides: insulin and amylin. Peptides 1998; 19 : 883–9. [Google Scholar]
- Rivera JF, Costes S, Gurlo T, et al. Autophagy defends pancreatic beta cells from human islet amyloid polypeptide-induced toxicity. J Clin Invest 2014; 124 : 3489–500. [Google Scholar]
- Hou X, Watzlawik JO, Fiesel FC, Springer W. Autophagy in Parkinson’s Disease. J Mol Biol 2020. [Google Scholar]
- Rivera JF, Gurlo T, Daval M, et al. Human-IAPP disrupts the autophagy/lysosomal pathway in pancreatic beta-cells: protective role of p62-positive cytoplasmic inclusions. Cell Death Differ 2011; 18 : 415–26. [Google Scholar]
- Winslow AR, Chen CW, Corrochano S, et al. alpha-Synuclein impairs macroautophagy: implications for Parkinson’s disease. J Cell Biol 2010; 190 : 1023–37. [Google Scholar]
- Masini M, Bugliani M, Lupi R, et al. Autophagy in human type 2 diabetes pancreatic beta cells. Diabetologia 2009; 52 : 1083–6. [Google Scholar]
- Costes S, Bertrand G, Ravier MA. Mechanisms of beta-cell apoptosis in type 2 diabetes-prone situations and potential protection by GLP-1-based therapies. Int J Mol Sci 2021; 22. [Google Scholar]
- Zeng H, Liu Y, Liu X, et al. Interplay of alpha-synuclein oligomers and endoplasmic reticulum stress in Parkinson’s disease: insights into cellular dysfunctions. Inflammation 2025; 48 : 1590–606. [Google Scholar]
- Xxxx S, Ahmad MH, Rani L, Mondal AC. Convergent Molecular pathways in type 2 diabetes mellitus and Parkinson’s disease: insights into mechanisms and pathological consequences. Mol Neurobiol 2022; 59 : 4466–87. [Google Scholar]
- Puspita L, Chung SY, Shim JW. Oxidative stress and cellular pathologies in Parkinson’s disease. Mol Brain 2017; 10 : 53. [Google Scholar]
- Zhang P, Zeng L, Gao W, et al. Peroxynitrite scavenger FeTPPS effectively inhibits hIAPP aggregation and protects against amyloid induced cytotoxicity. Int J Biol Macromol 2020; 161 : 336–44. [Google Scholar]
- Xu W, Huang Y, Zhou R. NLRP3 inflammasome in neuroinflammation and central nervous system diseases. Cell Mol Immunol 2025; 22 : 341–55. [Google Scholar]
- Groslambert M, Py BF. NLRP3, un inflammasome sous contrôle. Med Sci (Paris) 2018; 34 : 47–53. [Google Scholar]
- Lautier C, Grigorescu F. Maladies neurodégénératives et diabète. Med Sci (Paris) 2009; 25 : 337–40. [Google Scholar]
- Chen W, Cai W, Hoover B, Kahn CR. Insulin action in the brain: cell types, circuits, and diseases. Trends Neurosci 2022; 45 : 384–400. [Google Scholar]
- Rotermund C, Truckenmuller FM, Schell H, Kahle PJ. Diet-induced obesity accelerates the onset of terminal phenotypes in alpha-synuclein transgenic mice. J Neurochem 2014; 131 : 848–58. [Google Scholar]
- Lv YQ, Yuan L, Sun Y, et al. Long-term hyperglycemia aggravates alpha-synuclein aggregation and dopaminergic neuronal loss in a Parkinson’s disease mouse model. Transl Neurodegener 2022; 11 : 14. [Google Scholar]
- Nowotny K, Jung T, Hohn A, et al. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules 2015; 5 : 194–222. [Google Scholar]
- Hsu YH, Chen YW, Wu MH, Tu LH. Protein glycation by glyoxal promotes amyloid formation by islet amyloid polypeptide. Biophys J 2019; 116 : 2304–13. [Google Scholar]
- Abedini A, Cao P, Plesner A, et al. RAGE binds preamyloid IAPP intermediates and mediates pancreatic beta cell proteotoxicity. J Clin Invest 2018; 128 : 682–98. [Google Scholar]
- Vicente Miranda H, Szego EM, Oliveira LMA, et al. Glycation potentiates alpha-synuclein-associated neurodegeneration in synucleinopathies. Brain 2017; 140 : 1399–419. [Google Scholar]
- Armstrong MJ, Okun MS. Diagnosis and treatment of Parkinson disease: a review. JAMA 2020; 323 : 548–60. [Google Scholar]
- HAS. Haute autorité de santé: Stratégie thérapeutique du patient vivant avec un diabète de type 2. St-Denis : HAS, 2024. [Google Scholar]
- Phan F, Bertrand R, Amouyal C, Andreelli F. De la découverte des hormones incrétines aux agonistes doubles et triples du GIP / GLP-1 / glucagon. Med Sci (Paris) 2024; 40 : 837–47. [Google Scholar]
- Zaimia N, Obeid J, Varrault A, et al. GLP-1 and GIP receptors signal through distinct beta-arrestin 2-dependent pathways to regulate pancreatic beta cell function. Cell Rep 2023; 42 : 113326. [Google Scholar]
- Drucker DJ, Holst JJ. The expanding incretin universe: from basic biology to clinical translation. Diabetologia 2023; 66 : 1765–79. [Google Scholar]
- Harkavyi A, Abuirmeileh A, Lever R, et al. Glucagon-like peptide 1 receptor stimulation reverses key deficits in distinct rodent models of Parkinson’s disease. J Neuroinflammation 2008; 5 : 19. [Google Scholar]
- Li Y, Perry T, Kindy MS, et al. GLP-1 receptor stimulation preserves primary cortical and dopaminergic neurons in cellular and rodent models of stroke and Parkinsonism. Proc Natl Acad Sci USA 2009; 106 : 1285–90. [Google Scholar]
- Bertilsson G, Patrone C, Zachrisson O, et al. Peptide hormone exendin-4 stimulates subventricular zone neurogenesis in the adult rodent brain and induces recovery in an animal model of Parkinson’s disease. J Neurosci Res 2008; 86 : 326–38. [Google Scholar]
- Sun S, Huang L, Jiang G, et al. Neuroprotective effects of lixisenatide against propagation of alpha-synuclein pathology in Parkinson’s disease. Neural Regen Res 2025. [Google Scholar]
- Yun SP, Kam TI, Panicker N, et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat Med 2018; 24 : 931–8. [Google Scholar]
- Zhang LP, Zhang LY, Li L, Hölscher C. Semaglutide is neuroprotective and reduces α-synuclein levels in the chronic MPTP mouse model of Parkinson’s disease. J Parkinson Dis 2019; 9 : 157–71. [Google Scholar]
- Salameh TS, Rhea EM, Talbot K, Banks WA. Brain uptake pharmacokinetics of incretin receptor agonists showing promise as Alzheimer’s and Parkinson’s disease therapeutics. Biochem Pharmacol 2020; 180 : 114187. [Google Scholar]
- Wong CK, McLean BA, Baggio LL, et al. Central glucagon-like peptide 1 receptor activation inhibits Toll-like receptor agonist-induced inflammation. Cell Metab 2024; 36 : 130–43 e5. [Google Scholar]
- Zhang LY, Zhang LP, Li YW, et al. The novel dual GLP-1/GIP receptor agonist DA-CH5 is superior to single GLP-1 receptor agonists in the MPTP model of Parkinson’s disease. J Parkinson Dis 2020; 10 : 523–42. [Google Scholar]
- Zhang ZJ, Shi M, Li ZM, et al. A Dual GLP-1/GIP Receptor agonist is more effective than liraglutide in the A53T mouse model of Parkinson’s disease. Parkinsons Dis-Us 2023; 2023:7427136 [Google Scholar]
- Feng P, Liu Z, Lv D, et al. The novel GLP-1 / GIP dual agonist DA3-CH is more effective than liraglutide in the MPTP mouse model of Parkinson’s disease. Eur J Pharmacol 2025; 1003 : 177972. [Google Scholar]
- Zhang LY, Li C, Zhang ZJ, et al. DA5-CH and semaglutide protect against neurodegeneration and reduce a-synuclein levels in the 6-OHDA Parkinson’s disease rat model. Parkinsons Dis-Us 2022; 2022. [Google Scholar]
- Delvadia P, Dhote V, Mandloi AS, et al. Dual GLP-1 and GIP agonist tirzepatide exerted neuroprotective action in a Parkinson’s disease rat model. Acs Chemical Neuroscience 2025; 16 : 818–25. [Google Scholar]
- Tian R, Liu K, Lai H, et al. GLP-1/GIP dual agonist tirzepatide alleviates mice model of Parkinson’s disease by promoting mitochondrial homeostasis. Int Immunopharmacol 2025; 165 : 115443. [Google Scholar]
- Brauer R, Wei L, Ma T, et al. Diabetes medications and risk of Parkinson’s disease: a cohort study of patients with diabetes. Brain 2020; 143 : 3067–76. [Google Scholar]
- Gamborg M, Grand MK, Arvedsen J, et al. GLP-1 agonists as potential neuromodulators in development of Parkinson’s disease: a nationwide cohort study. Eur J Neurol 2025; 32 : e70075. [Google Scholar]
- Sun M, Wang X, Lu Z, et al. SGLT2 inhibitors vs. metformin for Parkinson’s disease risk reduction in type 2 diabetes. J Parkinsons Dis 2025 ; 15 :1240–1252. [Google Scholar]
- Kim HK, Biessels GJ, Yu MH, et al. SGLT2 inhibitor use and risk of dementia and Parkinson disease among patients with type 2 diabetes. Neurology 2024; 103 : e209805. [Google Scholar]
- Tang H, Lu Y, Okun MS, et al. Glucagon-like peptide-1 receptor agonists and risk of Parkinson’s disease in patients with type 2 diabetes: a population-based cohort study. Mov Disord 2024; 39 : 1960–70. [Google Scholar]
- Aviles-Olmos I, Dickson J, Kefalopoulou Z, et al. Exenatide and the treatment of patients with Parkinson’s disease. J Clin Invest 2013; 123 : 2730–6. [Google Scholar]
- Athauda D, Maclagan K, Skene SS, et al. Exenatide once weekly versus placebo in Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet 2017; 390 : 1664–75. [Google Scholar]
- Vijiaratnam N, Girges C, Auld G, et al. Exenatide once a week versus placebo as a potential disease-modifying treatment for people with Parkinson’s disease in the UK: a phase 3, multicentre, double-blind, parallel-group, randomised, placebo-controlled trial. Lancet 2025; 405 : 627–36. [Google Scholar]
- McGarry A, Rosanbalm S, Leinonen M, et al. Safety, tolerability, and efficacy of NLY01 in early untreated Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 2024; 23 : 37–45. [Google Scholar]
- Meissner WG, Remy P, Giordana C, et al. Trial of lixisenatide in early Parkinson’s disease. N Engl J Med 2024; 390 : 1176–85. [Google Scholar]
- Siddeeque N, Hussein MH, Abdelmaksoud A, et al. Neuroprotective effects of GLP-1 receptor agonists in neurodegenerative disorders: a large-scale propensity-matched cohort study. Int Immunopharmacol 2024; 143 : 113537. [Google Scholar]
Les îlots de Langerhans, ainsi nommés d’après le nom du biologiste allemand Paul Langerhans (1847-1888) qui les décrivit en 1868, sont des agrégats de cellules endocrines dont environ 60% sont des cellules β qui sécrètent l’insuline (ndlr).
La bradykinésie, ou lenteur, est un trouble moteur caractérisé par le ralentissement de mouvements volontaires, une difficulté à initier ou à exécuter les mouvements. Elle se manifeste par une réduction des mouvements automatiques, une diminution de l’expression faciale et des tâches quotidiennes (ndlr).
Le striatum ou néostriatum, appelé également le corps strié est une structure nerveuse subcorticale paire. Il est impliqué dans le mouvement involontaire, la motivation alimentaire ou sexuelle, la gestion de la douleur et la cicatrisation voire la régénérescence de certains tissus cérébraux. La pars compacta est l’une des parties de la substance noire (ndlr).
Les corps de Lewy, ainsi nommés d’après le neuroanatomiste allemand Frederic Lewy (1885-1950) qui a été le premier à découvrir les dépôts anormaux de protéines qui se forment à l’intérieur des cellules nerveuses au cours de la maladie de Parkinson, de la maladie à corps de Lewy et de certaines autres maladies neurodégénératives. Ils sont identifiés au microscope sur des coupes histologiques du cerveau (ndlr).
Les protéines intrinsèquement désordonnées (intrinsically disordered proteins, IDPs) sont très répandues dans le monde vivant. Elles sont composées de régions protéiques qui ne possèdent pas de structure tridimensionnelle fixe et stable dans des conditions physiologiques normales (ndlr).
Liste des figures
|
Figure 1 Liens entre diabète de type 2 et maladie de Parkinson. |
| Dans le texte | |
|
Figure 2 Évidences physiopathologiques et mécanistiques de l’association entre le diabète de type 2 et la maladie de Parkinson. L’agrégation des protéines amyloïdogéniques, IAPP et αS, dans leurs types cellulaires respectifs (et leur co-agrégation/cross-seeding dans les cellules β et les neurones), la résistance à l’insuline (centrale et/ou périphérique) ainsi que l’hyperglycémie conduisent à des mécanismes cellulaires communs d’altération : perturbation de l’autophagie, stress du réticulum endoplasmique, stress oxydant/mitochondrial et inflammation lesquels aboutissent à la dysfonction et/ou à la mort cellulaire. IAPP: islet amyloid polypeptide; αS: alpha-synucléine. |
| Dans le texte | |
|
Figure 3 Repositionnement thérapeutique des antidiabétiques GLP-1RA pour la maladie de Parkinson : évidences précliniques et cliniques. Les études précliniques réalisées chez les rongeurs montrent que l’utilisation des GLP-1RA entraîne une augmentation de la sécrétion d’insuline et une diminution de l’apoptose des cellules β pancréatiques et des neurones dopaminergiques par l’intermédiaire de certains mécanismes communs. Les GLP-1RA utilisés pour le traitement du diabète de type 2 favorisent une diminution de la résistance à l’insuline et de l’hyperglycémie. En ce qui concerne la maladie de Parkinson, les essais cliniques impliquant les GLP-1RA montrent un ralentissement de l’apparition et de la progression des troubles moteurs. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.