")
")
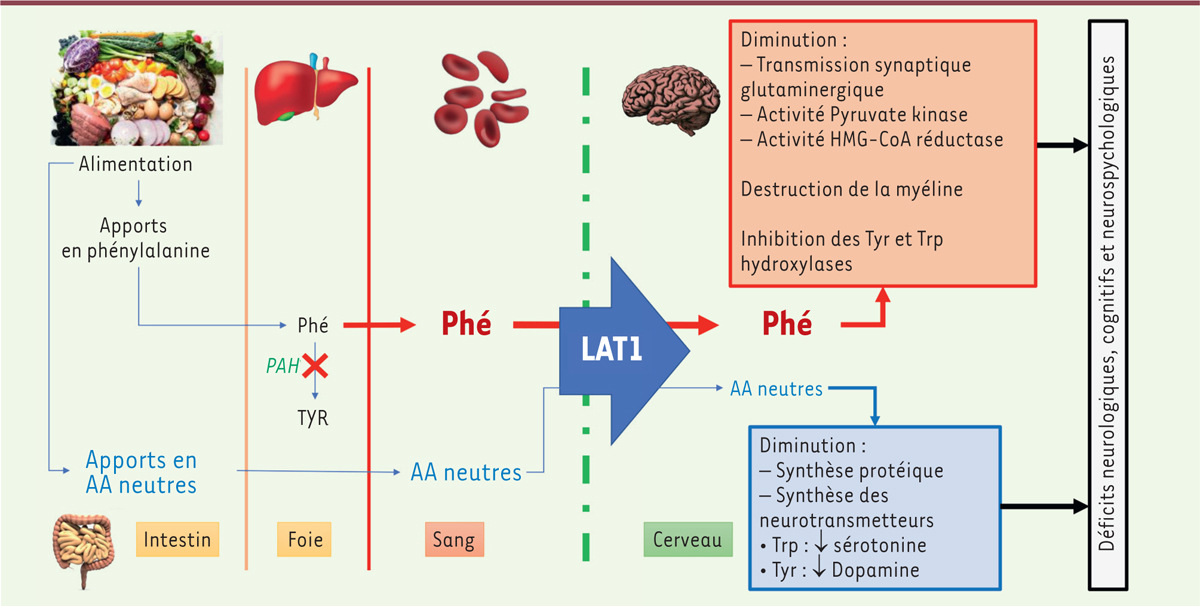
Figure 2.
Télécharger l'image originale
Physiopathologie de la phénylcétonurie. La phénylcétonurie est liée à un déficit de la dégradation de la phénylalanine (Phé), un acide aminé essentiel uniquement apporté par l’alimentation. Ce déficit entraîne une augmentation de la concentration plasmatique de Phé et une diminution de la synthèse de tyrosine (Tyr). La Phé et les acides aminés neutres (AAN) (tyrosine, tryptophane, thréonine, méthionine, valine, isoleucine, leucine, histidine) atteignent le cerveau via un même transporteur (LAT1, large neutral amino acid transporter) de façon compétitive. L’augmentation de la Phé plasmatique entraîne une augmentation de la Phé cérébrale qui a de multiples effets toxiques directs sur le métabolisme cérébral et entraîne également un déficit du passage des acides aminés neutres (qui sont tous, hormis la tyrosine, des acides aminés essentiels) au niveau cérébral. Ce déficit en AAN entraîne une baisse de la synthèse protéique intracérébrale et un déficit en neurotransmetteurs dépendants de la tyrosine et du tryptophane (Trp). Ces anomalies conjuguées entraînent les déficits neurologiques cognitifs et neuropsychologiques de la PCU.
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.