")
")
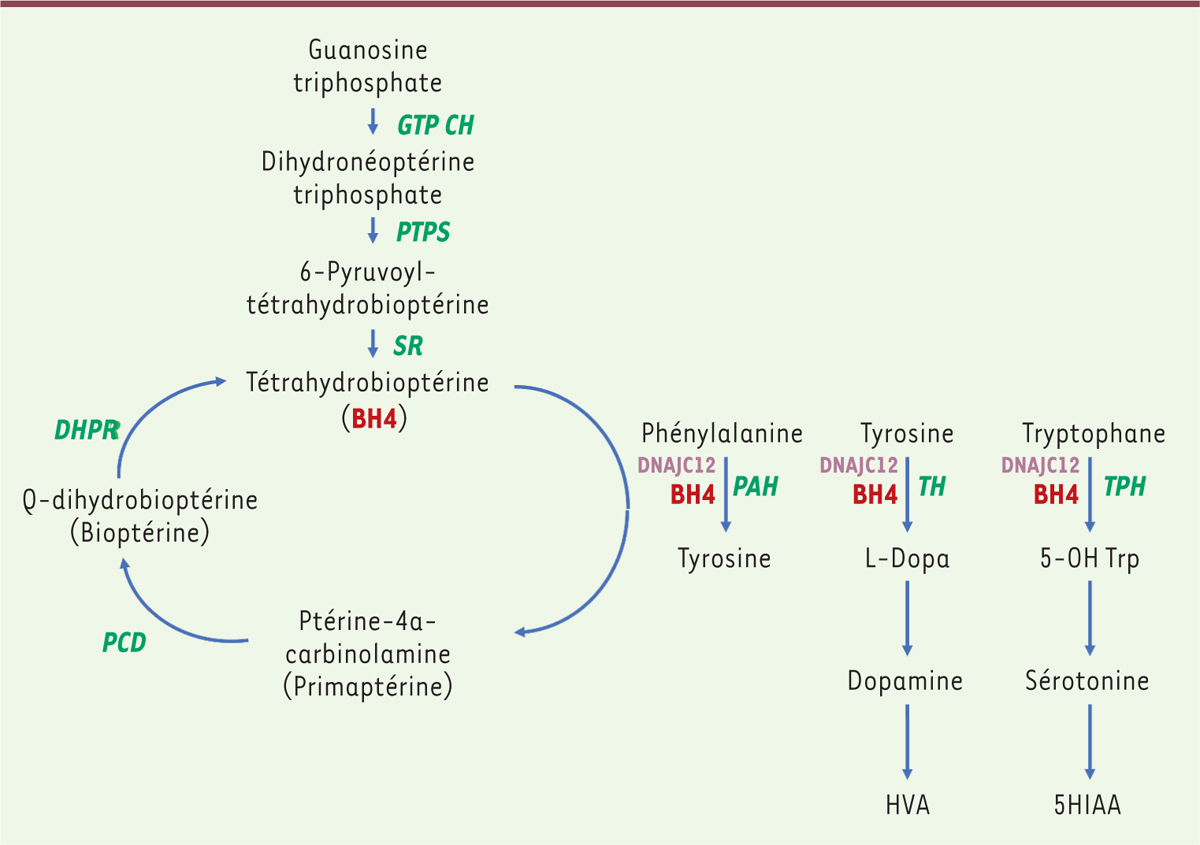
Figure 1.
Télécharger l'image originale
Voie métabolique de la phénylcétonurie. La phénylcétonurie est liée à un déficit de la phénylalanine hydroxylase (PAH) dont le fonctionnement nécessite la présence d’un cofacteur, la tétrahydrobioptérine (BH4). Ce cofacteur est également nécessaire au fonctionnement de la tyrosine hydroxylase (TH) et de la tryptophane hydroxylase (TPH). Une insuffisance de PAH entraîne une hyperphénylalaninémie isolée. Une réduction de la synthèse du BH4 (liée à des déficits en guanosine triphosphate cyclohydrolase [GTPCH], pyruvoyl-tétrahydrobioptérine synthase [PTPS] et sépiaptérine réductase [SR]) ou de son recyclage (lié au déficit en ptérine-4a-carbinolamine déhydratase [PCD] et dihydroptéridine réductase [DHPR]) seront à l’origine d’une hyperphénylalaninémie mais également d’un déficit en neurotransmetteurs. Une molécule chaperonne (DNAJC12) est également nécessaire au bon fonctionnement de ces trois hydroxylases. L’acide homovanillique (HVA) et l’acide 5-hydroxylindolacétique (5HIAA) sont deux métabolites issus de la voie de la dopamine et de la sérotonine.
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.