")
")
| Issue |
Med Sci (Paris)
Volume 34, Novembre 2018
Les Cahiers de Myologie
|
|
|---|---|---|
| Page(s) | 26 - 31 | |
| Section | Fiche pratique | |
| DOI | https://doi.org/10.1051/medsci/201834s208 | |
| Published online | 12 novembre 2018 | |
La myopathie à agrégats tubulaires et le syndrome de Stormorken
Tubular aggregate myopathy and Stormorken syndrome
1
Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC), Illkirch, France
2
Centre National de la Recherche Scientifique (CNRS), UMR7104, Illkirch, France
3
Institut National de la Santé et de la Recherche Médicale (INSERM), U1258, Illkirch, France
4
Université de Strasbourg, Illkirch, France
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Le calcium est un régulateur essentiel pour un grand nombre de fonctions cellulaires, et une perturbation de l’homéostasie calcique peut sévèrement troubler la physiologie de différents tissus. CASQ1, STIM1, et ORAI1 codent pour des facteurs clés contrôlant les flux de calcium, et des mutations de ces gènes sont à l’origine de la myopathie à agrégats tubulaires et du syndrome de Stormorken. Ces deux maladies forment un continuum clinique regroupant faiblesse musculaire, myosis, thrombopénie, hyposplénisme, ichthyose, dyslexie et petite taille.
Abstract
Calcium (Ca2+) is an essential regulator for a large number of cellular functions in various tissues and organs, and small disturbances of Ca2+ homeostasis can severely compromise normal physiology. Intracellular Ca2+ balance is mainly controlled by the reticular Ca2+ sensor STIM1 and the plasma membrane Ca2+ channel ORAI1 through a mechanism known as store-operated Ca2+ entry (SOCE). Gain-of-function mutations in STIM1 or ORAI1 cause excessive extracellular Ca2+ influx, resulting in tubular aggregate myopathy (TAM) and Stormorken syndrome (STRMK). Both disorders are spectra of the same disease and involve muscle weakness, miosis, thrombocytopenia, hyposplenism, ichthyosis, dyslexia, and short stature. Here we summarize the clinical and histological characteristics of both disorders, provide an overview on the genetic causes, and recapitulate the current knowledge on the pathomechanisms leading to the multi-systemic phenotype of tubular aggregate myopathy and Stormorken syndrome.
© 2018 médecine/sciences – Inserm
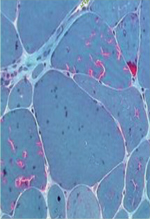
La myopathie à agrégats tubulaires (TAM) est une maladie musculaire progressive caractérisée par une faiblesse musculaire, des crampes et myalgies, et les biopsies de personnes atteintes montrent typiquement des agencements denses de tubules membranaires [1]. Certains patients présentent des signes cliniques additionnels comme myosis, thrombocytopénie, hyposplénisme, ichthyose, dyslexie, et petite taille, et ce phénotype multi-systémique est appelé syndrome de Stormorken (STRMK) [2].
La myopathie à agrégats tubulaires et syndrome de Stormorken sont dus à des mutations hétérozygotes de STIM1 [3–6] ou ORAI1 [6, 7], alors que des mutations hétérozygotes de CASQ1 ont été trouvées chez des patients avec un phénotype exclusivement musculaire [8, 9].
Les trois gènes codent des régulateurs clés de l’homéostasie calcique, et agissent de ce fait sur une multitude de voies cellulaires dépendantes du calcium, dont la contraction musculaire. La calséquestrine, codée par CASQ1, joue un rôle majeur dans le stockage de calcium dans le réticulum, et participe activement au relargage du calcium vers le cytosol [10]. STIM1 est un senseur réticulaire de calcium, et peut activer le canal calcique ORAI1, situé dans la membrane plasmique, afin d’induire l’entrée de calcium extracellulaire à travers un mécanisme appelé SOCE (store-operated calcium entry) [11–13]. Le SOCE existe dans tout type de cellule, ce qui sous-tend le large spectre phénotypique chez les patients TAM/STRMK avec mutations de STIM1 ou ORAI1. Des analyses fonctionnelles ont démontré qu’il s’agit de mutations gain-de-fonction provoquant une sur-activation du SOCE et une entrée excessive de calcium [3, 6, 7].
Cet article vise à résumer les caractéristiques cliniques et histologiques de la myopathie à agrégats tubulaires et du syndrome de Stormorken, et à analyser la corrélation entre génotype et phénotype avec une attention particulière pour les pathomécanismes entraînant le dysfonctionnement musculaire.
Présentation clinique et histologique de la myopathie à agrégats tubulaires et du syndrome de Stormorken
Au niveau clinique, la myopathie à agrégats tubulaires se fait surtout remarquer par des crampes, des myalgies ainsi qu’une faiblesse musculaire progressive touchant d’abord les muscles proximaux des membres inférieurs [1]. Le niveau de créatine kinase dans le sérum est souvent dix fois supérieur à la normale, et certains patients développent des rétractions dans les bras et les jambes, ainsi que des troubles de l’oculomotricité [3, 7, 9, 14]. Des symptômes multi-systémiques comme une thrombocytopénie, un myosis, une ichthyose, une dyslexie, ou une petite taille peuvent être plus ou moins prononcés, et la totalité de cette présentation clinique est diagnostiqué en tant que syndrome de Stormorken [2, 7, 15–19]. De manière générale, TAM et STRMK forment un continuum clinique avec des patients TAM qui peuvent montrer un ou plusieurs signes du syndrome de Stormorken, et certains patients avec syndrome de Stormorken qui présentent surtout une faiblesse musculaire [16]. À noter qu’une troisième maladie fait également partie de ce continuum : le syndrome plaquettaire de York (YPS) a initialement été décrit comme maladie hématologique [20], mais des examens cliniques complets ont révélé des symptômes musculaires et non-musculaires correspondant à la TAM et au syndrome de Stormorken [16].
Les biopsies musculaires de patients TAM/STRMK montrent des accumulations basophiles centrales ou sous-sarcolemmiques surtout dans les fibres de type II. Ces accumulations apparaissent en rouge après coloration de trichrome de Gomori, et en bleu après coloration NADH-TR. A l’échelle ultra-structurale, ces accumulations correspondent à des agencements réguliers de tubules membranaires d’un diamètre de 20 à 200 nm [1, 21] (Figure 1A). À part ce trait caractéristique, d’autres signes histopathologiques comme une variabilité de taille de fibres, des noyaux internalisés, une prédominance de fibres de type I, ou une atrophie des fibres de type II ont également été décrits chez de patients avec TAM/STRMK [3, 7, 9, 14, 15, 17, 18, 22, 23].
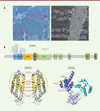 |
Figure 1. A.Coloration de trichrome de Gomori d’une biopsie musculaire d’un patient TAM/STRMK montrant une inégalité de taille de fibres, ainsi que des accumulations basophiles en rouge. La microscopie électronique dévoile que ces accumulations correspondent à des agencements réguliers de tubules membranaires. B. STIM1 est une protéine transmembranaire du réticulum avec les domaines EF et SAM formant la partie luminale, un domaine transmembranaire (TM), et des domaines coiled-coil (CC1, CC2, CC3) contenant le SOAR (STIM1-ORAI1 activating domain), un domaine inhibiteur (ID), et des régions riches en sérine/proline (SP) et lysine (K) dans la partie cytosolique. La majorité des mutations faux-sens s’accumule dans les domaines EF liant le calcium. ORAI1 est un canal calcique de la membrane plasmique, et chaque monmère est composé de quatre domaines transmembranaires (hélices alpha colorées) avec le domaine M1 formant le puit. La position des mutations est marquée en rouge. La calséquestrine (CASQ1) est composée de trois domaines thiorédoxines globulaires avec un noyau hydrophobe et une surface d’acides aminés négativement chargés pour lier le calcium. Les mutations sont marquées en rouge à l’exception d’I385T, qui n’apparaît pas sur le modèle de la structure résolue de 383 acides aminés. |
Origine des agrégats tubulaires et présence dans d’autres myopathies
Les agrégats tubulaires ne sont pas réactifs aux colorations histologiques SDH et COX, ce qui exclut une origine mitochondriale [1, 24]. Des analyses sur coupes musculaires par immunofluorescence ont démontré que les agrégats tubulaires contiennent différentes protéines réticulaires comme le senseur de calcium STIM1, les pompes calciques SERCA1 et SERCA2, le canal calcique RyR1, et le tampon de calcium calséquestrine [1, 3, 7, 9, 15, 23]. Il est donc probable qu’il s’agit de structures issues du réticulum sarcoplasmique. Cependant, le mécanisme de formation des agrégats tubulaires n’est pas connu. Il est possible que l’entrée excessive de calcium dans les fibres musculaires de patients avec TAM ou syndrome de Stormorken induise une dilatation du réticulum en tant que compartiment principal de stockage de calcium, et que des replis membranaires dus à cette dilatation représentent une première étape dans la formation des agrégats tubulaires.
Il est important de noter que les agrégats tubulaires sont aussi trouvés en tant que traits secondaires dans d’autres maladies musculaires transmises ou acquises telles que l’hyperthermie maligne, la paralysie périodique hypokaliémique, ou les myopathies métaboliques, inflammatoires et éthyliques [25–27]. Les agrégats tubulaires sont également présents dans les syndromes myasthéniques dus à des mutations de DPAGT1, ALG2, ou encore GFPT1 [28].
Gènes et protéines impliqués
La majorité des patients TAM/STRMK avec diagnostic moléculaire porte des mutations de STIM1, et les mutations d’ORAI1 sont moins fréquentes. Il s’agit toujours de mutations hétérozygotes avec gain-de fonction, alors que des mutations récessives perte-de-fonction dans ces deux gènes ont été associées à une immunodéficience sévère, caractérisée par des infections récurrentes et chroniques, une auto-immunité, une dysplasie ectodermale, et une hypotonie musculaire [29–31]. CASQ1 a été récemment identifié comme troisième gène de TAM chez des patients qui ne manifestent aucun des signes non-musculaires du syndrome de Stormorken [8, 9].
STIM1 est une protéine transmembranaire avec une partie N-terminale dans la lumière du réticulum, et une partie C-terminale cytosolique. La partie luminale contient deux domaines EF capables de lier le calcium, et un domaine SAM (sterile a-motif), resserré aux domaines EF à l’état inactif [32]. La déplétion des réserves calciques et le détachement du calcium des domaines EF induit un changement de conformation et un dépliement de STIM1, qui va ensuite di- et multimériser [32]. La partie cytosolique de STIM1 est constituée de différents domaines et régions, dont notamment le domaine SOAR (STIM1-ORAI1 activating domain) permettant l’interaction avec le canal calcique ORAI1 et son activation, et une hélice inhibitrice régulant cette interaction [11, 33, 34].
ORAI1 fait partie des canaux CRAC (calcium release-activated calcium channels), qui se distinguent par une forte sélectivité pour le calcium, et une faible conductance [35]. ORAI1 est activé suite à la déplétion des réserves calciques par STIM1 et le SOCE, et inactivé par des concentrations élevées de calcium cytosolique (CDI ; calcium-dependent inactivation). Ce canal de la membrane plasmique est composé de tétramères ou hexamères qui forment des anneaux concentriques autour d’un puit central [36–38]. Chaque sous-unité contient quatre domaines transmembranaires (M1 à M4), avec M1 formant le puit, et M2 à M4 les anneaux [39]. L’ouverture du canal est régulé par l’interaction des parties N- et C-terminales avec STIM1 [40].
Contrairement à l’expression ubiquitaire de STIM1 et ORAI1, la calséquestrine est uniquement retrouvée dans le muscle squelettique, et notamment dans les fibres rapides de type II [41]. La calséquestrine est une protéine globulaire avec noyau hydrophobe et une surface exposant des acides aminés acides permettant la liaison électrostatique du calcium [42]. Elle participe activement au relargage de calcium vers le cytosol dans un complexe quartenaire avec RyR1, la junctine, et la triadine [10]. Elle est monomérique à faible concentration calcique, et les monomères de calséquestrine séquestrent STIM1 et agissent ainsi en tant que régulateurs négatifs du SOCE [43]. L’entrée de calcium dans le réticulum induit la di- et polymérisation de la calséquestrine, ce qui augmente sa capacité de liaison de calcium [44–46].
Corrélation génotype/phénotype
À présent, 14 mutations différentes de STIM1 ont été rapportées chez des patients TAM/STRMK, dont 12 mutations des domaines EF (H72Q, N80T, G81D, D84E, D84G, S88G, L96V, F108I, F108L, H109N, H109R, I115F) [3, 14, 17, 18, 23], et deux mutations de la partie cytosolique (R304Q et R304W, Figure 1B) [4–6, 16, 23, 47]. Il s’agit exclusivement de mutations faux-sens touchant des acides aminés conservés. La majorité de ces mutations est unique ; N80T, G81D, et H109N ont été trouvées deux fois, et H109R et I115F trois fois. La mutation R304W dans la partie cytosolique est la plus récurrente avec 12 familles connues et non-apparentées. L’autre mutation cytosolique touchant le même acide aminé, R304Q, a été rapportée dans une seule famille [23]. Il existe une corrélation partielle entre le génotype et le phénotype. La mutation récurrente R304W a principalement été trouvée chez des patients avec phénotype multi-systémique de syndrome de Stormorken [4–6, 23, 47], et seuls quelques patients ont principalement une gêne musculaire [16]. À l’inverse, les patients avec mutation des domaines EF présentent surtout un phénotype musculaire et peu ou pas de symptômes non-musculaires. Le phénotype du muscle squelettique se traduit par une faiblesse musculaire ou une myalgie comme signe primaire et majeur [3, 14, 16–18, 22, 23], et cela ne dépend pas uniquement de l’acide aminé affecté. La substitution du résidu F108 en isoleucine cause une faiblesse musculaire proximale dès l’enfance dans une famille, et le changement en leucine est associé à une myalgie à début tardif dans une autre famille [14]. Il existe même une variabilité clinique pour la même mutation. Une famille avec la mutation H109N était décrite avec fatigabilité post-exercice, et une autre famille avec la même mutation montrait une faiblesse musculaire et des rétractions des membres inférieurs [3, 14].
Les mutations d’ORAI sont moins fréquentes avec sept familles TAM/STRMK rapportées à présent [6, 7, 15, 19]. Toutes les mutations touchent des acides aminés conservés dans les domaines transmembranaires : les faux-sens S97C, G98S, et V107M ont été trouvées dans le domaine transmembranaire M1 constituant le puit, et L138F, T184M, et P245L dans les domaines formant les anneaux concentriques (Figure 1B). G98S est la seule mutation rapportée deux fois, et les deux familles manifestent un phénotype sévère avec faiblesse musculaire marquée et rétractions dès l’enfance [7, 15]. En revanche, la famille avec la mutation voisine S97C était cliniquement plus modérée avec crampes et faiblesse musculaire à début tardif [19]. Généralement, on observe des crampes et une raideur musculaire surtout chez les patients ORAI1 [6, 7, 15, 19, 48], et moins chez les patients STIM1. Une autre différence concerne les symptômes non-musculaires, qui sont globalement moins prononcés chez les patients ORAI1, et certains signes du syndrome de Stormorken comme la thrombocytopénie, l’asplénie, ou la petite taille n’ont jamais été rapportés. La faiblesse musculaire est plutôt diffuse en cas de mutation d’ORAI1, et souvent proximale pour STIM1. Des études d’IRM et de tomographie des membres inférieurs de patients ORAI1 ont démontré une atrophie symétrique et des infiltrations graisseuses dans les parties postérieures et médiales de la cuisse, et de la partie postérieure du bas de la jambe avec implication particulière des muscles gastrocnémiens [7, 15, 19]. Les patients STIM1 montrent une atrophie et des infiltrations graisseuses dans la cuisse et le bas de jambe à l’exception de quelques muscles comme le tibial antérieur [49]. À noter que le muscle long fléchisseur de l’hallux, rarement affecté dans les maladies musculaires, est atteint chez les patients avec mutations de STIM1 et ORAI1 [19, 49], ce qui pourrait représenter un signe distinctif entre la myopathie à agrégats tubulaires et le syndrome de Stormorken.
CASQ1 a récemment été identifié en tant que troisième gène TAM, et quatre mutations hétérozygotes différentes ont été décrites : D44N, N56Y, G103D, et I385T (Figure 1B) [8, 9]. Le phénotype des patients est modéré par rapport aux patients STIM1 et ORAI1, et se limite à une faiblesse musculaire des membres inférieurs peu progressive et à début tardif, ainsi qu’à des myalgies et une fatigue post-exercice [8, 9, 50]. Ceci est en accord avec l’expression exclusivement musculaire de CASQ1 [41], qu’on trouve surtout dans les fibres de types II. Une autre mutation faux-sens dans CASQ1 (D244G) a été préalablement associée à la myopathie vacuolaire, qui diffère de la myopathie à agrégats tubulaires au niveau clinique et histologique [51].
Pathomécanismes des mutations STIM1/ORAI1/CASQ1
Dans des cellules transfectées, STIM1 a une localisation réticulaire diffuse, alors que l’expression de constructions avec mutations des domaines EF induit l’oligomérisation constitutive de la protéine [3, 14, 17, 18, 22]. Ceci suggère que les mutations du domaine EF impactent directement ou indirectement la coordination du calcium ou déstabilisent l’interaction entre les domaines EF et SAM, et provoquent ainsi le dépliement et l’oligomérisation de STIM1, et en conséquence l’activation du canal calcique ORAI1 sans déplétion préalable des réserves calciques. En effet, des analyses ratiométriques sur myoblastes de patients ont révélé un niveau calcique basal élevé et une entrée excessive de calcium extracellulaire sans activation du SOCE [3, 18]. La mutation cytosolique R304W a le même effet dans les cellules transfectées et dans les myoblastes de patients [4–6, 23], mais le pathomécanisme est différent. Une étude récente a démontré que cette mutation induit une élongation hélicale dans le domaine cytosolique de STIM1 provoquant l’exposition du domaine SOAR et l’activation d’ORAI1 [52]. Cette même étude a révélé un effet moins délétère de la mutation R304Q, ce qui va de pair avec un phénotype plus modéré des patients respectifs. L’effet pléiotropique de la mutation R304W peut s’expliquer par une répercussion particulière sur l’inactivation d’ORAI1 calcium-dépendante (CDI), comme démontré par des études électrophysiologiques [6]. Comparée aux mutations des domaines EF, la mutation R304W provoque une entrée calcique prolongée en conséquence d’une répression plus intense du CDI [6], et c’est ce surplus de calcium dans différents cellules et tissus qui serait à l’origine de symptômes multi-systémiques du syndrome de Stormorken.
L’expression exogène des mutants ORAI1 induit une augmentation du niveau calcique basal et une entrée excessive de calcium après activation du SOCE [7, 15, 19], et des résultats similaires ont été obtenus dans des myotubes de patients [15, 19]. Des études dans des cellules murines sans Stim1 et son paralogue Stim2 (Stim1-/-/Stim2-/-) ont été effectuées par la suite afin de déterminer si l’abondance de calcium était le résultat d’une perméabilité accrue ou d’une activation anormale du canal [7]. Une entrée constante d’ions était uniquement observée dans des cellules exprimant ORAI1 avec des mutations du domaine M1 (G98S et V107M), alors qu’ORAI1 avec des mutations du domaine M3 (T184M) était indiscernable du contrôle. En revanche, les trois mutations induisaient bien une entrée excessive de calcium dans des cellules Stim1-/-/Stim2-/- co-exprimant ORAI1 et STIM1. Ces résultats indiquent un pathomécanisme différent en fonction des mutations. Les mutations touchant des acides aminés du puit génèrent un canal perméable indépendamment de STIM1, alors que des mutations des anneaux concentriques causent une activation anormale du canal via STIM1 et le SOCE [6, 7].
Des études dans cellules transfectées et sur protéines recombinantes ont montré que les mutations de CASQ1 entravaient la dynamique de polymérisation et dépolymérisation de la calséquestrine [8, 9]. Les mutants monomérisaient moins que le contrôle après déplétion des réserves calciques, et produisaient moins de grands polymères à des concentrations calciques croissantes. Ces observations suggèrent un double effet pathogénique des mutations de CASQ1. Dans ce modèle, il y aurait moins de monomères de calséquestrine pour séquestrer STIM1 et inhiber le SOCE, et moins de polymères de calséquestrine pour assurer le stockage de calcium dans le réticulum. En effet, le traitement de fibres musculaires isolées à la caféine a confirmé une capacité de stockage de calcium diminué dans le réticulum [8]. Contrairement aux mutations TAM, le mutant D244G, auparavant associé à la myopathie vacuolaire [51], produit des grands polymères insolubles de calséquestrine. Ceci suggère que les mutations de CASQ1 peuvent être à l’origine de deux myopathies différentes, et que ces deux maladies impliquent un pathomécanisme différent [8, 9].
Résumé et directions futures
Pour résumer, des mutations hetérozygotes gain-de-fonction dans STIM1 et ORAI1 induisent une entrée excessive de calcium et causent la myopathie à agrégats tubulaires et le syndrome de Stormorken [3–7], tandis que les mutations hétérozygotes de CASQ1 promeuvent l’entrée de calcium à travers un impact sur la polymérisation et dépolymérisation de la calséquestrine [8, 9]. À l’inverse, des mutations récessives perte-de-fonction de STIM1 et ORAI1 suppriment l’entrée de calcium et entraînent une immunodéficience [29–31]. En conclusion, TAM et STRMK sont causés par un SOCE sur-actif, et l’immunodéficience par un SOCE sous-actif.
La recherche ultérieure sur la myopathie à agrégats tubulaires et le syndrome de Stormorken devra aborder des questions fondamentales sur la physiopathologie de ces maladies avec une attention particulière aux effets en aval de l’entrée excessive de calcium afin de déterminer la séquence des événements qui conduisent au dysfonctionnement musculaire et aux anomalies dans d’autres tissus. Même si l’excès de calcium est indubitablement à l’origine des phénotypes majeurs, il reste à déterminer si un déséquilibre potentiel d’autres ions contribue au développement de la maladie. Il est par exemple possible que les mutations d’ORAI1 impactent sur la sélectivité ionique du canal, et que la sur-activation de STIM1 due à des mutations influence d’autres canaux. Il est en effet connu que STIM1 régule les canaux TRP (transient potential receptor), qui sont bien moins sélectifs pour le calcium [53].
Il n’y a actuellement pas de traitement pour la myopathie à agrégats tubulaires et le syndrome de Stormorken. Néanmoins, le déséquilibre de calcium est susceptible à la manipulation par des agents pharmacologiques, et la réduction de l’entrée excessive de calcium à travers des molécules inhibant le canal calcique ORAI1 pourraient représenter une approche thérapeutique adaptée.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Remerciements
Les auteurs souhaitent remercier Raphaël Schneider pour son aide pour la réalisation de la figure.
Références
- Chevessier F Bauche-Godard S Leroy JP et al. The origin of tubular aggregates in human myopathies. J Pathol 2005 ; 207 : 313–323. [CrossRef] [PubMed] [Google Scholar]
- Stormorken H Sjaastad O Langslet A et al. A new syndrome: thrombocytopathia, muscle fatigue, asplenia, miosis, migraine, dyslexia and ichthyosis. Clin Genet 1985 ; 28 : 367–374. [PubMed] [Google Scholar]
- Bohm J Chevessier F Maues De Paula A et al. Constitutive activation of the calcium sensor STIM1 causes tubular-aggregate myopathy. Am J Hum Genet 2013 ; 92 : 271–278. [Google Scholar]
- Misceo D Holmgren A Louch WE et al. A dominant STIM1 mutation causes Stormorken syndrome. Hum Mutat 2014 ; 35 : 556–564. [CrossRef] [PubMed] [Google Scholar]
- Morin G Bruechle NO Singh AR et al. Gain-of-function mutation in STIM1 (P.R304W) is associated with Stormorken syndrome. Hum Mutat 2014 ; 35 : 1221–1232. [CrossRef] [PubMed] [Google Scholar]
- Nesin V Wiley G Kousi M et al. Activating mutations in STIM1 and ORAI1 cause overlapping syndromes of tubular myopathy and congenital miosis. Proc Natl Acad Sci USA 2014 ; 111 : 4197–4202. [CrossRef] [Google Scholar]
- Bohm J Bulla M Urquhart JE et al. ORAI1 mutations with distinct channel gating defects in tubular aggregate myopathy. Hum Mutat 2017 ; 38 : 426–438. [CrossRef] [PubMed] [Google Scholar]
- Barone V Del Re V Gamberucci A et al. Identification and characterization of three novel mutations in the CASQ1 gene in four patients with tubular aggregate myopathy. Hum Mutat 2017 ; 38 : 1761–1773. [CrossRef] [PubMed] [Google Scholar]
- Bohm J Lornage X Chevessier F et al. CASQ1 mutations impair calsequestrin polymerization and cause tubular aggregate myopathy. Acta Neuropathol 2018 ; 135 : 149–151. [CrossRef] [PubMed] [Google Scholar]
- Lee KW Maeng JS Choi JY et al. Role of Junctin protein interactions in cellular dynamics of calsequestrin polymer upon calcium perturbation. J Biol Chem 2012 ; 287 : 1679–1687. [CrossRef] [PubMed] [Google Scholar]
- Park CY Hoover PJ Mullins FM et al. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 2009 ; 136 : 876–890. [CrossRef] [PubMed] [Google Scholar]
- Luik RM Wu MM Buchanan J et al. The elementary unit of store-operated Ca2+ entry: local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J Cell Biol 2006 ; 174 : 815–825. [CrossRef] [PubMed] [Google Scholar]
- Stathopulos PB Zheng L Li GY et al. Structural and mechanistic insights into STIM1-mediated initiation of store-operated calcium entry. Cell 2008 ; 135 : 110–122. [CrossRef] [PubMed] [Google Scholar]
- Bohm J Chevessier F Koch C et al. Clinical, histological and genetic characterisation of patients with tubular aggregate myopathy caused by mutations in STIM1. J Med Genet 2014 ; 51 : 824–833. [CrossRef] [PubMed] [Google Scholar]
- Endo Y Noguchi S Hara Y et al. Dominant mutations in ORAI1 cause tubular aggregate myopathy with hypocalcemia via constitutive activation of store-operated Ca2+ channels. Hum Mol Genet 2015 ; 24 : 637–648. [CrossRef] [PubMed] [Google Scholar]
- Markello T Chen D Kwan JY et al. York platelet syndrome is a CRAC channelopathy due to gain-of-function mutations in STIM1. Mol Genet Metab 2015 ; 114 : 474–482. [Google Scholar]
- Noury JB Bohm J Peche GA et al. Tubular aggregate myopathy with features of Stormorken disease due to a new STIM1 mutation. Neuromuscul Disord 2017 ; 27 : 78–82. [CrossRef] [PubMed] [Google Scholar]
- Walter MC Rossius M Zitzelsberger M et al. 50 years to diagnosis: Autosomal dominant tubular aggregate myopathy caused by a novel STIM1 mutation. Neuromuscul Disord 2015 ; 25 : 577–584. [CrossRef] [PubMed] [Google Scholar]
- Garibaldi M Fattori F Riva B et al. A novel gain-of-function mutation in ORAI1 causes late-onset tubular aggregate myopathy and congenital miosis. Clin Genet 2017 ; 91 : 780–786. [CrossRef] [PubMed] [Google Scholar]
- White JG Giant electron-dense chains, clusters and granules in megakaryocytes and platelets with normal dense bodies: an inherited thrombocytopenic disorder. Platelets 2003 ; 14 : 109–121. [Google Scholar]
- Muller HD Vielhaber S Brunn A et al. Dominantly inherited myopathy with novel tubular aggregates containing 1–21 tubulofilamentous structures. Acta Neuropathol 2001 ; 102 : 27–35. [PubMed] [Google Scholar]
- Hedberg C Niceta M Fattori F et al. Childhood onset tubular aggregate myopathy associated with de novo STIM1 mutations. J Neurol 2014 ; 261 : 870–876. [CrossRef] [PubMed] [Google Scholar]
- Harris E Burki U Marini-Bettolo C et al. Complex phenotypes associated with STIM1 mutations in both coiled coil and EF-hand domains. Neuromuscul Disord 2017 ; 27 : 861–872. [CrossRef] [PubMed] [Google Scholar]
- Chevessier F Marty I Paturneau-Jouas M et al. Tubular aggregates are from whole sarcoplasmic reticulum origin: alterations in calcium binding protein expression in mouse skeletal muscle during aging. Neuromuscul Disord 2001 ; 14 : 208–216. [Google Scholar]
- Schiaffino S. Tubular aggregates in skeletal muscle: just a special type of protein aggregates?. Neuromuscul Disord 2012 ; 22 : 199–207. [CrossRef] [PubMed] [Google Scholar]
- Goebel HH When tubules aggregate. Neuromuscul Disord 2012 ; 22 : 208–210. [CrossRef] [PubMed] [Google Scholar]
- Engel WK Bishop DW Cunningham GG Tubular aggregates in type II muscle fibers: ultrastructural and histochemical correlation. J Ultrastruct Res 1970 ; 31 : 507–525. [Google Scholar]
- Engel AG Shen XM Selcen D et al. Congenital myasthenic syndromes: pathogenesis, diagnosis, and treatment. Lancet Neurol 2015 ; 14 : 420–434. [CrossRef] [PubMed] [Google Scholar]
- Feske S Gwack Y Prakriya M et al. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006 ; 441 : 179–185. [CrossRef] [PubMed] [Google Scholar]
- Picard C McCarl CA Papolos A et al. STIM1 mutation associated with a syndrome of immunodeficiency and autoimmunity. N Engl J Med 2009 ; 360 : 1971–1980. [Google Scholar]
- Lacruz RS Feske S Diseases caused by mutations in ORAI1 and STIM1. Ann NY Acad Sci 2015 ; 1356 : 45–79. [CrossRef] [Google Scholar]
- Stathopulos PB Li GY Plevin MJ et al. Stored Ca2+ depletion-induced oligomerization of stromal interaction molecule 1 (STIM1) via the EF-SAM region: an initiation mechanism for capacitive Ca2+ entry. J Biol Chem 2006 ; 281 : 35855–35862. [CrossRef] [PubMed] [Google Scholar]
- Yuan JP Zeng W Dorwart MR et al. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat Cell Biol 2009 ; 11 : 337–343. [CrossRef] [PubMed] [Google Scholar]
- Kawasaki T Lange I Feske S A minimal regulatory domain in the C terminus of STIM1 binds to and activates ORAI1 CRAC channels. Biochem Biophys Res Commun 2009 ; 385 : 49–54. [Google Scholar]
- Prakriya M. The molecular physiology of CRAC channels. Immunol Rev 2009 ; 231 : 88–98. [CrossRef] [PubMed] [Google Scholar]
- Cai X Zhou Y Nwokonko RM et al. The Orai1 store-operated calcium channel functions as a hexamer. J Biol Chem 2016 ; 291 : 25764–25775. [CrossRef] [PubMed] [Google Scholar]
- Hou X Pedi L Diver MM et al. Crystal structure of the calcium release-activated calcium channel Orai. Science 2012 ; 338 : 1308–1313. [Google Scholar]
- Thompson JL Shuttleworth TJ How many Orai’s does it take to make a CRAC channel?. Sci Rep 2013 ; 3 : 1961. [CrossRef] [PubMed] [Google Scholar]
- Vig M Beck A Billingsley JM et al. CRACM1 multimers form the ion-selective pore of the CRAC channel. Curr Biol 2006 ; 16 : 2073–2079. [CrossRef] [PubMed] [Google Scholar]
- Zheng H Zhou MH Hu C et al. Differential roles of the C and N termini of Orai1 protein in interacting with stromal interaction molecule 1 (STIM1) for Ca2+ release-activated Ca2+ (CRAC) channel activation. J Biol Chem 2013 ; 288 : 11263–11272. [CrossRef] [PubMed] [Google Scholar]
- Schiaffino S Reggiani C Fiber types in mammalian skeletal muscles. Physiol Rev 2011 ; 91 : 1447–1531. [Google Scholar]
- Wang S Trumble WR Liao H et al. Crystal structure of calsequestrin from rabbit skeletal muscle sarcoplasmic reticulum. Nat Struct Biol 1998 ; 5 : 476–483. [CrossRef] [PubMed] [Google Scholar]
- Wang L Zhang L Li S et al. Retrograde regulation of STIM1-Orai1 interaction and store-operated Ca2+ entry by calsequestrin. Sci Rep 2015 ; 5 : 11349. [CrossRef] [PubMed] [Google Scholar]
- Park H Park IY Kim E et al. Comparing skeletal and cardiac calsequestrin structures and their calcium binding: a proposed mechanism for coupled calcium binding and protein polymerization. J Biol Chem 2004 ; 279 : 18026–18033. [CrossRef] [PubMed] [Google Scholar]
- Park H Wu S Dunker AK et al. Polymerization of calsequestrin. Implications for Ca2+ regulation. J Biol Chem 2003 ; 278 : 16176–16182. [CrossRef] [PubMed] [Google Scholar]
- Cho JH Ko KM Singaruvelu G et al. Functional importance of polymerization and localization of calsequestrin in C. elegans. J Cell Sci 2007 ; 120 : 1551–1558. [Google Scholar]
- Alonso-Jimenez A Ramon C Dols-Icardo O et al. Corpus callosum agenesis, myopathy and pinpoint pupils: consider Stormorken syndrome. Eur J Neurol 2018 ; 25 : e25–e26. [CrossRef] [PubMed] [Google Scholar]
- Shahrizaila N Lowe J Wills A Familial myopathy with tubular aggregates associated with abnormal pupils. Neurology 2004 ; 63 : 1111–1113. [Google Scholar]
- Tasca G D’Amico A Monforte M et al. Muscle imaging in patients with tubular aggregate myopathy caused by mutations in STIM1. Neuromuscul Disord 2015 ; 25 : 898–903. [CrossRef] [PubMed] [Google Scholar]
- Rohkamm R Boxler K Ricker K et al. A dominantly inherited myopathy with excessive tubular aggregates. Neurology 1983 ; 33 : 331–336. [Google Scholar]
- Rossi D Vezzani B Galli L et al. A mutation in the CASQ1 gene causes a vacuolar myopathy with accumulation of sarcoplasmic reticulum protein aggregates. Hum Mutat 2014 ; 35 : 1163–1170. [CrossRef] [PubMed] [Google Scholar]
- Fahrner M Stadlbauer M Muik M et al. A dual mechanism promotes switching of the Stormorken STIM1 R304W mutant into the activated state. Nat Commun 2018 ; 9 : 825. [CrossRef] [PubMed] [Google Scholar]
- Worley PF Zeng W Huang GN et al. TRPC channels as STIM1-regulated store-operated channels. Cell Calcium 2007 ; 42 : 205–211. [CrossRef] [PubMed] [Google Scholar]
Liste des figures
|
Figure 1. A.Coloration de trichrome de Gomori d’une biopsie musculaire d’un patient TAM/STRMK montrant une inégalité de taille de fibres, ainsi que des accumulations basophiles en rouge. La microscopie électronique dévoile que ces accumulations correspondent à des agencements réguliers de tubules membranaires. B. STIM1 est une protéine transmembranaire du réticulum avec les domaines EF et SAM formant la partie luminale, un domaine transmembranaire (TM), et des domaines coiled-coil (CC1, CC2, CC3) contenant le SOAR (STIM1-ORAI1 activating domain), un domaine inhibiteur (ID), et des régions riches en sérine/proline (SP) et lysine (K) dans la partie cytosolique. La majorité des mutations faux-sens s’accumule dans les domaines EF liant le calcium. ORAI1 est un canal calcique de la membrane plasmique, et chaque monmère est composé de quatre domaines transmembranaires (hélices alpha colorées) avec le domaine M1 formant le puit. La position des mutations est marquée en rouge. La calséquestrine (CASQ1) est composée de trois domaines thiorédoxines globulaires avec un noyau hydrophobe et une surface d’acides aminés négativement chargés pour lier le calcium. Les mutations sont marquées en rouge à l’exception d’I385T, qui n’apparaît pas sur le modèle de la structure résolue de 383 acides aminés. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.