")
")
| Issue |
Med Sci (Paris)
Volume 32, Number 1, Janvier 2016
Origine développementale de la santé et des maladies (DOHaD), environnement et épigénétique
|
|
|---|---|---|
| Page(s) | 27 - 34 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/20163201006 | |
| Published online | 5 février 2016 | |
Le nouveau paradigme de l’origine développementale de la santé et des maladies (DOHaD)
Épigénétique, environnement : preuves et chaînons manquants
The new paradigm of the developmental origin of health and diseases (DOHaD) – Epigenetics and environment: evidence and missing links
1
Inra, UMR1198, biologie du développement et reproduction, Domaine de Vilvert, Bâtiment 230, F-78352 Jouy-en-Josas, France
2
Université Pierre et Marie Curie, F-75005 Paris, France
3
Institut Carmen, Inserm U1060, Oullins, France
4
UMR 1280 Inra université de Nantes, Institut des maladies de l’appareil digestif, Nantes, France
5
Inserm UMR1037, Centre de recherche en cancérologie de Toulouse, université de Toulouse III Paul Sabatier, F-31037 Toulouse, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Selon le nouveau paradigme des origines développementales de la santé et des maladies (DOHaD), des modifications épigénétiques retiennent la mémoire des effets des facteurs environnementaux auxquels un individu est soumis tout au long de sa vie. Une période cruciale est celle du développement précoce, pré- et postnatal ; cruciale parce que l’épigénome y est particulièrement sensible aux effets de l’environnement, et aussi parce que l’individu construit son capital santé pour répondre ultérieurement, plus ou moins bien, aux aléas de la vie. Le défi pour la recherche est de déchiffrer comment les facteurs conférant une susceptibilité accrue ou une résilience vis-à-vis du développement des maladies agissent et influencent les mécanismes épigénétiques. Le défi pour la puissance publique est de traduire en actions et en bénéfices pour la santé ces découvertes scientifiques via, entre autres, l’établissement de recommandations capables de freiner l’incidence croissante des maladies chroniques.
Abstract
According to the new paradigm of the Developpemental Origins of Health and Disease (DOHaD), the environmental factors to which an individual is exposed throughout his life can leave an epigenetic footprint on the genome. A crucial period is the early development, where the epigenome is particularly sensitive to the effects of the environment, and during which the individual builds up his health capital that will enable him to respond more or less well to the vagaries of life. The research challenge is to decipher the modes of action and the epigenetic mechanisms put into play by environmental factors that lead to increased disease susceptibility or resilience. The challenge for health is to translate these scientific discoveries into action through, among others, the establishment of preventive recommendations to slow down the growing incidence of non communicable diseases.
© 2016 médecine/sciences – Inserm

Épigénétique et environnement : la révolution
La découverte de l’incorporation des impacts de l’environnement dans l’épigénome représente une véritable révolution : qui aurait pu imaginer que les épreuves physiques et psychologiques endurées par des femmes enceintes lors de l’épisode Ice storm au Québec en 19981, ou des attentats du 11 septembre 20012 allaient laisser des traces dans l’épigénome des fœtus et influencer leur phénotype et leur devenir [1, 2] ? Ainsi, selon le nouveau paradigme des « origines développementales de la santé et des maladies (DOHaD) », l’environnement peut laisser sur notre épigénome des traces qui sont conservées longtemps après l’exposition à l’événement, voire qui sont transmises aux générations suivantes, sans altérer la séquence de l’ADN, via nos épigénomes. Dès 2004, les groupes de M. Szyf et de M. Meaney montrèrent que, chez le rat, la qualité des soins maternels durant la période postnatale conditionne pour la vie la réactivité au stress de leur progéniture [3]. Ces soins modifient, dans l’hippocampe, l’épigénome d’un gène clé, celui qui code le récepteur aux glucocorticoïdes, mais également l’épigénome de réseaux de gènes [4]. Des mécanismes semblables opèrent chez le rongeur, le macaque et l’humain [5, 6], montrant la conservation entre les espèces des mécanismes impliqués dans de tels processus d’adaptation à l’environnement. Des mécanismes épigénétiques ont depuis été impliqués dans les effets, sur la progéniture, de divers stimulus environnementaux subis à différents stades par la mère ou par le père : nutrition déséquilibrée, adversité, stress, peur du prédateur, défaite sociale, etc. [5, 7–12] (→).
(→) Voir la Synthèse de M.A. Charles et al., page 15 de ce numéro
Ces découvertes remettent en question la séparation entre l’inné et l’acquis, très prisée aux siècles derniers, au profit d’une frontière fluctuante et virtuelle. Le séquençage du génome a apporté une connaissance fabuleuse, mais ne nous offre qu’un nombre restreint de marqueurs de prédiction fiables quant à la susceptibilité à certaines maladies chroniques, une application qui n’est pas à la hauteur de l’espoir suscité. Une étude récente estime qu’à la naissance, les interactions gène/environnement liées à des processus épigénétiques représentent 75 % de la variabilité phénotypique, alors que le génotype ne rend compte que de 25 % [13]. Et pourtant, les marques épigénétiques ne s’apposent pas au hasard, mais dépendent aussi de la séquence de l’ADN. Il est donc nécessaire de mieux comprendre les relations entre génétique et épigénétique, de les associer plutôt que de les opposer ou d’ignorer l’une ou l’autre [10, 14].
Selon l’OMS (Organisation mondiale de la santé), la prévalence des maladies chroniques augmentera de 17 % au cours de la prochaine décennie, ce qui représentera un coût humain et financier énorme. Le concept de DOHaD, parce qu’il s’intéresse aux origines de pathologies observées chez l’adulte (en général chroniques) et ne se limite pas à une intervention à la période où la maladie est déjà installée – ce qui limite les chances de succès de la combattre – offre des possibilités de prévention. Il permet de distinguer, en termes d’impact environnemental et de réponse, trois grandes phases couvrant le cycle de la vie dans son intégralité (Figure 1). Nous ne développerons ici que les deux premières phases, la troisième faisant l’objet de la seconde revue de C. Junien et al. (→).
(→) Voir la Synthèse de C. Junien et al., page 35 de ce numéro
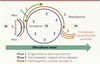 |
Figure 1. Le cycle de la vie. La DOHaD comprend trois grandes phases : (1) la phase développementale de programmation de l’individu – couvrant la gestation, la petite enfance, et aussi l’adolescence – caractérisée par une grande plasticité de l’épigénome ; (2) la phase des effets à moyen et long terme, conséquences des pertur- bations développementales, mais auxquelles viennent s’associer, tout au long de la vie, les effets fluctuants ou permanents de l’environnement ; (3) la phase de reproduction puis les effets ou réponses inter- et trans- générationnels. Au cours de ces phases, les différentes perturbations subies par les marques épigénétiques sous l’influence de l’environnement modulent le phénotype. Bien que le terme « transgénérationnel » puisse être uti- lisé souvent indifféremment, on se doit de distinguer les effets transgénérationnels des effets intergénérationnels ou multigénérationnels. Les premiers impliquent une transmission par la lignée germinale, pour la première génération hors de tout contact avec l’agent environnemental perturbateur. Cet effet transgénérationnel ne peut apparaître qu’au niveau de la F3 par la lignée maternelle, ou de la F2 par la lignée paternelle. Les effets observés en deçà sont appelés intergénérationnels. Les effets multigénérationnels correspondent à une exposition au même stress ou à un stress qui peut découler de la première exposition, sur plusieurs généra- tions. Le point rouge représente, après la conception, le début de la gamétogenèse : le terme naissance reste postérieur. Enfin, il existe des différences entre les mâles et les femelles dues, dès la conception, à la différence entre les paires de chromosomes sexuels (XX et XY) puis, après la différenciation des gonades, à l’expression des hormones sexuelles. Des différences très précoces d’expression de gènes de la machinerie épigénétique apposent des marques épigénétiques spécifiques du sexe qui établissent de façon temporaire ou pérenne des réseaux fonctionnels de gènes différents chez le mâle et la femelle. La gamétogenèse et la résistance à la reprogrammation sont aussi différentes entre le mâle et la femelle. On observe ainsi un dimor- phisme sexuel, tant au niveau basal que pour les réponses à court terme ou à long terme [30]. |
Pour comprendre les enjeux de ce nouveau concept de la DOHaD, il est essentiel de définir des termes comme épigénétique, dynamique versus stable, héritabilité au sens mitotique ou transgénérationnel, héréditaire, ainsi que les limites pratiques de la réversibilité théorique des marques épigénétiques. Ces définitions permettront d’expliciter les sources de confusion et de montrer comment l’épigénétique, un des supports moléculaires des phénomènes observés mais probablement pas le seul, offre dans toute sa diversité des possibilités insoupçonnées pour comprendre les processus de la DOHaD. Dans cette synthèse, notre objectif n’est pas de faire une revue exhaustive des mécanismes épigénétiques. D’excellentes revues décrivent les différents acteurs et les multiples variations épigénétiques observées jusqu’à présent dans diverses conditions et différentes espèces [15–19]. Notre but est de montrer, à l’aide de travaux récents, les vraies questions, approches et stratégies transdisciplinaires qui restent à mettre en place pour aborder ces mécanismes dans le contexte de la DOHaD (Figure 2).
 |
Figure 2. Génétique et épigénétique. Notre patrimoine génétique composé de 3 milliards de paires de bases, identique dans toutes nos cellules, n’est pas nu dans le noyau : il entoure un complexe protéique formé de 4 paires d’histones formant une série de « perles » appelées nucléosomes. Des modifications réversibles appo- sées soit sur la séquence d’ADN (méthylation), soit sur les histones, participent au degré de compaction de la chromatine permettant ainsi à un gène de s’exprimer ou à l’inverse d’être réprimé. Ces mécanismes sont impliqués dans toute une série de mécanismes dont la transcription (expression), la réparation-répli- cation, la condensation, l’inactiva- tion de l’X, l’empreinte parentale, le vieillissement et le dimorphisme sexuel, etc. De plus, contrairement aux marques génétiques qui sont irréversibles, les marques épigénétiques, sensibles à tout type d’environnement, sont, par nature, réversibles. |
Les définitions de l’épigénétique : éphémères, ambiguës et controversées
En 1942, Conrad H. Waddington inventait le terme « épigénétique » (en anglais, epigenetics), compilation des termes « epigenesis » et « genetics » ; celui-ci désignait « la branche de la biologie qui étudie les relations de cause à effet entre les gènes et leurs produits, faisant apparaître le phénotype »3 [20]. Le sens du terme épigénétique change ensuite grâce à l’élucidation progressive de ses fondements moléculaires et devient « l’étude des changements de la fonction des gènes héritables au niveau mitotique et/ou méiotique qui ne peuvent être expliqués par des changements dans la séquence de l’ADN »4 [21]. La première définition couvre l’activité des biologistes du développement, la seconde laisse la porte ouverte à tout ce qui n’est pas mutation dans l’ADN.
Puis Adrian Bird a revu la définition pour proposer : « l’adaptation structurelle de régions chromosomiques permettant d’enregistrer, de signaler ou de perpétuer des états d’activité modifiés »5 [22]. Plus récemment, l’étude de systèmes, comme celui de l’horloge circadienne, qui par définition permet de faire fluctuer l’expression des gènes au cours des 24 h de la journée [23], ou, comme la mémoire neuronale [24], révèle l’implication de mécanismes et d’outils typiquement épigénétiques mais n’impliquant, ni l’un ni l’autre, de division cellulaire, donc d’héritabilité mitotique [22]. Selon Adrian Bird, exiger le critère « héritable » pour définir un mécanisme comme étant épigénétique, c’est-à-dire la capacité de s’autoperpétuer lors de la division cellulaire, est source de confusions, d’amalgames, surtout avec la notion populaire de transmission à la génération suivante ; il ne se justifie plus et devrait être abandonné [22].
Phase 1 – Ontogenèse des paysages épigénétiques : dynamique et/ou stabilité de la chromatine
L’épigénome est constitué de multiples strates et facettes interconnectées qui varient au cours du temps et selon l’environnement. Après la fécondation, l’épigénome des gamètes parentaux subit une reprogrammation drastique : l’enlèvement des marques épigénétiques caractéristiques des gamètes et l’acquisition de nouvelles marques sont essentiels pour assurer la totipotence nécessaire au développement. Les mécanismes épigénétiques dans l’embryon précoce comprennent la méthylation, l’hydroxyméthylation de l’ADN et les modifications post-traductionnelles des histones (→) et peuvent également inclure le remplacement de variants d’histones par d’autres. Dans les cellules souches embryonnaires (embryonic stem cells, ES), un réseau unique de facteurs de transcription appelés facteurs pionniers (OCT4 [octamer-binding transcription factor 4], SOX2 [sex determining region Y)-box 2] et NANOG [Homeobox protein NANOG], pour les principaux) régule l’établissement et la maintenance de la pluripotence de ces cellules. L’introduction de ces facteurs pionniers dans une cellule souche totipotente, mais aussi dans une cellule somatique différenciée, entraîne des modifications épigénétiques très spécifiques (reprogrammation) qui changent de manière spectaculaire le phénotype d’une cellule [25, 26]. Les profils des marques épigénétiques mises en place successivement au cours du développement permettent de représenter dans toute leur diversité les programmes d’expression génique nécessaires pour construire harmonieusement les différentes parties d’un organisme [27]. Ainsi nos 23 000 gènes ne s’expriment pas de la même façon dans les différents types cellulaires et selon le sexe ou l’âge. Cette plasticité dépend de marques épigénétiques versatiles et également d’ARN non codants [28] pour répondre à un environnement qui change, à tous les stades de la vie [29].
(→) Voir la Synthèse de C. Junien et al., page 35 de ce numéro
Pourquoi les phases précoces du développement constituent des fenêtres sensibles ou insensibles
Les phases précoces du développement offrent une large gamme d’opportunités éphémères pour façonner les marques épigénétiques, la conformation de la chromatine, au gré de l’environnement, selon le stade de développement, l’âge, le statut physiopathologique, le sexe et, plus tard, le genre [30]. Les cellules ES6 ont une chromatine « ouverte » et active. Les gènes de développement sont marqués à la fois par la marque épigénétique répressive H3K27me3 (correspondant à la triméthylation de la lysine 27 de l’histone H3) et par la marque activatrice H3K4me3 (triméthylation de la lysine 4 de l’histone H3) d’où le terme de marques bivalentes. Ces gènes sont silencieux mais « prêts » à s’exprimer (primed) ou à être inhibés, selon qu’ils perdent l’une ou l’autre des deux marques avant ou au cours de la différenciation cellulaire, qui s’accompagne d’un remodelage global de la chromatine [31] (Figure 3). Les cellules ES éteignent les gènes de pluripotence et acquièrent le phénotype de cellules différenciées distinctes en activant les gènes spécifiques de lignage et en réprimant les gènes spécifiques des autres lignages inappropriés [31]. Ainsi les gènes actifs contiennent généralement la marque « activatrice » H3K4me3 au niveau de leur promoteur et les marques H3K4me1/me2 et H3K27ac (lysine 27 acétylée) au niveau de leurs enhancers. Les locus réprimés sont enrichis en H3K27me3 et/ou en méthylation de l’ADN, qui semblent réprimer des locus distincts. Il n’est donc pas étonnant qu’un facteur environnemental puisse laisser un impact dans un contexte chromatinien donné et pas dans un autre.
 |
Figure 3. Héritabilité mitotique : de quoi la chromatine se souvient-elle et comment ? Mécanismes moléculaires sous-tendant la transmission épigénétique mitotique. Le schéma montre la réplication de l’ADN (bleu foncé, ADN parental ; bleu clair, ADN nouvellement synthétisé) à la fourche de réplication (flèche orange). Lors de la réplication, les nucléosomes parentaux (gris) sont dissociés et réassemblés sur les brins d’ADN néoformés en utilisant des histones parentales (gris) et nouvellement synthétisées (bleu). Les marques d’histone sont illustrées sous forme de carrés verts et les mécanismes responsables de leur transmission sont encadrées en vert. La méthylation de l’ADN est illustrée sous forme de cercles rouges et les mécanismes responsables de la main- tenance de la méthylation sont encadrées en rouge. La transmission mitotique des marques épigénétiques peut être dépendante de la réplication, par exemple via le couplage avec la fourche de réplication, ou indépendante de la machinerie de réplication. Actuellement, on connaît très peu de choses sur la façon dont les domaines de la chromatine active sont maintenus (tracé vert clair). Contrairement à la fidélité de recopiage de la séquence d’ADN et de la méthylation de l’ADN, la duplication des histones ne se fait pas à partir d’une matrice et les modifications post-traductionnelles ne sont pas conservées au moment où la cellule se divise. Les marques d’histones semblent s’apposer conformément à l’état transcriptionnel, mais après la réplication. Les gènes sont activés par des protéines régulatrices, les facteurs de transcription (FT), qui se fixent à l’ADN au niveau des promoteurs et enhancers. Au cours de la réplication cellulaire, ceux-ci sont distribués aux cellules filles qui les séquestrent car leur présence continuelle est requise. Les histones néosynthétisées viennent s’associer aux histones parentales porteuses des modifications post-traductionnelles, disloquées lors de la progression de la fourche de répli- cation. Des protéines de type chaperon les accompagnent. Contrairement à la méthylation de l’ADN, le mécanisme par lequel les profils de modifications post-traductionnelles des histones sont rétablis après chaque division demeure obscur et l’objet d’ardents débats. En effet, des altérations des systèmes Polycomb/trithorax sont souvent héritées par les générations suivantes de cellules et parfois d’organismes (d’après [61]). |
Comment garder la mémoire ? L’héritabilité mitotique
Quels mécanismes assurent la propagation de la mémoire des impacts de l’environnement ? Chez les eucaryotes, hormis durant les processus de différenciation, le statut actif ou réprimé des gènes est héritable puisqu’il est transmis à l’identique au cours des divisions cellulaires, à la descendance des cellules [32].
L’héritabilité n’a donc pas le même sens selon que l’on parle d’héritabilité « mitotique » – au cours des divisions cellulaires d’un même organisme –, ce qui faisait partie de la définition moderne initiale, ou d’héritabilité « transgénérationnelle », d’observation plus récente, et qui fait intervenir une transmission via les gamètes. Les mécanismes ou les agents potentiellement impliqués, qu’ils soient épigénétiques ou autres (ARN non codants, facteurs diffusibles ou cytoplasmiques, par exemple) sont différents dans ces deux types d’héritabilité. De plus, bien que certaines marques épigénétiques soient considérées comme généralement stables, des influences environnementales et des événements stochastiques peuvent être à l’origine de modifications de ces marques et changer le statut transcriptionnel [33].
Jusqu’à présent, l’attention s’était surtout portée sur les 2 principaux codes épigénétiques : les modifications enzymatiques des histones et la méthylation de l’ADN (Figure 3). Cette dernière est fidèlement reproduite lors de la division, par la méthyltransférase 1 de l’ADN (DNMT1) [34], et peut donc être le support de la mémoire. Cependant, certains organismes comme la drosophile ou le nématode Caenorhabditis elegans, n’ont pas d’enzyme permettant la méthylation de l’ADN, mais sont capables, par d’autres mécanismes, d’auto-perpétuer des états d’expression altérés. En revanche, chez tous les eucaryotes, il existe bien une batterie d’enzymes capables d’apporter des modifications post-traductionnelles aux histones. Les systèmes Polycomb/Trithorax, par leur activité méthyltransférase sur les histones, sont également des acteurs importants (Figure 3).
Il existe donc différents processus capables de conserver la mémoire des impacts de l’environnement. Il est probable qu’ils n’agissent pas seuls mais en interaction les uns avec les autres et avec d’autres processus qui restent à découvrir.
Réversibilité des marques ou réversibilité des états phénotypiques ?
Si, par nature, les marques épigénétiques sont malléables donc réversibles, les conséquences de leurs changements sur la morphogenèse ou l’état de la cellule peuvent, elles, être irréversibles. Au cours des périodes critiques de plasticité développementale (préconceptionnelle, gestation, petite enfance et adolescence), un environnement subnormal peut créer une prédisposition irréversible aux facteurs de risque d’une maladie survenant ultérieurement chez l’adulte. Ainsi, une alimentation déséquilibrée de la mère pendant la gestation peut être associée, chez l’enfant, à une diminution du nombre de cellules bêta pancréatiques productrices d’insuline, ou de néphrons, entraînant respectivement un risque de diabète ou d’hypertension (→).
(→) Voir la Synthèse de A. Gabory et al., page 66 de ce numéro
Une adversité précoce peut, elle, exposer à un risque de maladie neuro-dégénérative ou de développement tumoral [35–37] (→).
(→) Voir la Synthèse de C. Delpierre et al., page 21 de ce numéro
La dynamique inhérente aux processus épigénétiques explique l’importante flexibilité des marques en fonction de l’exposition à des facteurs de l’environnement internes et externes [38]. Si certaines, établies lors de l’impact, sont stables, d’autres apparaîtront ou/et disparaîtront, faisant évoluer le paysage épigénétique tout en aggravant ou en corrigeant le phénotype au cours du temps [39] (Figure 4).
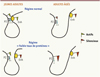 |
Figure 4. La mémoire est dynamique à travers l’es- pace, boucle, promoteurs, enhancers. Modèle simpli- fié proposé pour la boucle formée par l’association entre l’enhancer (Enh, activateur transcriptionnel) et le promoteur (P2) dans le locus Hnf4a (hepatocyte nuclear factor-4-alpha) dans les îlots pancréatiques d’animaux contrôles jeunes et âgés (rangée du haut) et dénutris en protéines (rangée du bas). Dans les îlots des jeunes rats adultes contrôles, l’enhancer (Enh) est enrichi en marques d’histone actives et interagit étroitement avec le promoteur P2 de Hnf4a, conduisant à des niveaux élevés de transcription de P2. L’interaction de l’enhancer-promoteur avec P2 est altérée dans les îlots des animaux exposés à une alimentation pauvre en protéines au cours du développement précoce. Ceci est dû à l’épuisement en marques d’histones activatrices et à l’enrichisse- ment en marques d’histones inhibitrices au niveau de la région de l’enhancer. Le vieillissement est associé à une réduction des niveaux d’expression d’Hnf4a en raison de l’enrichissement en marques d’histones inhibitrices le long du locus, et ceci est plus prononcé dans les îlots des animaux LP. L’accumulation de marques d’histones inhibitrices pourrait affaiblir davantage l’interaction de l’enhancer- promoteur avec P2. La différence entre les adultes contrôles et les adultes tests dans la figure se manifeste par un écartement de la base de la boucle entre le promoteur et l’enhancer, un écartement qui est déjà prononcé entre les jeunes adultes soumis à un régime normal par rapport aux adultes âgés. La différence entre les 2 régimes s’accroît chez les adultes âgés soumis à une exposition précoce au régime pauvre en protéine (d’après [39]). |
Pourtant, à l’inverse des mutations du génome, les multiples épimutations (mutations de l’épigénome) sont, elles, potentiellement réversibles. Les facteurs épigénétiques étalent leurs effets sur la structure de la chromatine sur des échelles de temps variées allant de la minute, au cours de la signalisation par un récepteur, à des années pour la mémoire métabolique, ou à des générations pour les perturbations environnementales [38, 40]. Ainsi, l’ADN subit des cycles de méthylation/déméthylation indiquant qu’une marque « stable » d’activité génique peut toutefois être modulée par des facteurs de transcription et répondre dynamiquement à des signaux de l’environnement [41].
Contrairement à la séquence génétique, unique et stable dans toutes les cellules, les profils épigénétiques varient d’un type cellulaire à l’autre au sein d’un même tissu, voire même de la partie apicale à la partie distale d’un même tissu. Ils sont dynamiques au cours de la vie [42] et des rythmes circadiens [23]. De fait, l’élément central de l’horloge circadienne, le facteur de transcription CLOCK (clock circadian regulator), possède également une activité acétyltransférase d’histone et plusieurs modificateurs de la chromatine sont recrutés de manière circadienne au niveau des promoteurs des gènes contrôlés par l’horloge ; des ARN antisens jouent également un rôle important dans cette régulation [43, 62]. Il est maintenant également admis que des processus épigénétiques sous-tendent les états fonctionnels des neurones qui peuvent rester stables pendant plusieurs années. Toutefois, ces états ne pourront être transmis à des cellules filles, puisqu’à part quelques exceptions, les neurones ne se divisent pas [24, 44].
S’il existe aujourd’hui au moins une vingtaine d’exemples de correction ou de compensation d’effets transmis par la mère qui influençent le phénotype de la progéniture [39, 45], un petit nombre seulement fait état de modifications épigénétiques accompagnant ces changements phénotypiques au cours de la vie d’un individu [46–48]. Il n’existe cependant pas encore, à notre connaissance, d’exemple de réversibilité de marques associées à des effets correcteurs ou compensateurs au cours des générations.
Phase 2 : comment l’organisme programmé répond à l’environnement
La seconde phase de la DOHaD correspond aux effets, ou plutôt aux réponses, à moyen et long terme, aux perturbations survenues au cours du développement, auxquelles viennent s’associer celles survenues tout au long de la vie, sous l’influence d’effets permanents et fluctuants de l’environnement (Figure 1).
Le principe des deux événements : effets ou réponses ?
Les altérations épigénétiques survenues au cours du développement sous les effets de l’environnement peuvent être considérées comme un premier événement. Elles ne confèrent qu’un état latent, une sensibilité à un second événement, révélé plus tard par des facteurs de risque environnementaux. Plus qu’un effet causal direct à long terme, il s’agit plutôt de modifications de la capacité de réponse des tissus/organes programmés [36, 37, 49].
Les marques évoluent rapidement au cours du temps [39, 42]. Les changements ultérieurs qui surviennent sous l’effet de différents environnements, ou des modifications liées au vieillissement, peuvent augmenter ou diminuer la charge allostatique c’est-à-dire rendre un individu encore plus susceptible, ou inversement, plus résistant, à leurs effets (Figure 4) [39].
La « programmation » précoce peut également conférer à un gène une capacité à répondre plus rapidement à une seconde exposition. Ainsi, le concept de mémoire hépatique suggère qu’un gène (par exemple les gènes codant les récepteurs aux hormones stéroïdes et thyroïdes) peut répondre plus rapidement à un inducteur lors de la deuxième exposition que lors de l’activation initiale. Cela est particulièrement évident si celle-ci a lieu à un stade où cet inducteur n’est habituellement pas présent, et la mémoire de cette exposition peut être retenue pendant plusieurs mois [50]. Ainsi, cela pourrait expliquer pourquoi certains polluants aux effets hormono-mimétiques (perturbateurs endocriniens) ont des effets avant la différenciation des gonades, donc malgré l’absence d’hormones gonadiques à ce stade (→).
(→) Voir la Synthèse de C. Mauduit et al., page 45 de ce numéro
La mémoire métabolique : peut-on la déverrouiller ?
On ne sait pas si toutes les marques épigénétiques acquises sont réversibles et dans quelles conditions. Chez l’humain, un des mécanismes les mieux étudiés d’interaction entre un défaut nutritionnel et l’épigénome est la mémoire métabolique ou glycémique, observée chez des patients diabétiques (→).
(→) Voir le numéro thématique Diabète : approches thérapeutiques émergentes, m/s n°8-9, août-septembre 2013
Chez ces patients, les complications du diabète causées par l’hyperglycémie transitoire persistent voire progressent, alors même que la glycémie est contrôlée par l’administration d’insuline [51]. L’analyse des modifications post-traductionnelles des histones dans des cellules sanguines de patients atteints de diabète de type 1 (DT1) a montré que l’acétylation de la lysine 9 de l’histone H3 (H3K9ac) est modifiée [52]. De façon similaire, in vitro dans des cellules endothéliales primaires exposées à une hyperglycémie, cette acétylation de l’histone H3 est diminuée sur un nombre significatif de régions promotrices de gènes impliqués dans la physiopathologie du DT1 [53].
Ces observations indiquent qu’une exposition à des concentrations élevées de glucose induit des modifications épigénétiques stables qui pourraient expliquer la mémoire métabolique chez l’humain. Compte tenu du caractère réversible des marques épigénétiques, il reste cependant à déterminer pourquoi, et surtout, comment, ces marques sont-elles « verrouillées », puisqu’un retour à une glycémie normale ne les efface pas ?
Dans des cellules mammaires normales, l’exposition aux estrogènes induit la formation transitoire de multiples boucles dans une région du chromosome 16 qui rapprochent 14 locus distants. Les points d’interactions, se focalisant sur des sites sensibles aux œstrogènes, permettent ainsi une répression coordonnée de ces locus. Cependant, dans les cellules mammaires tumorales, l’acquisition d’une méthylation de l’ADN et de modifications post-traductionnelles des histones ayant une action répressive aboutit à un échafaudage inflexible. Du fait de cette perte de flexibilité, la répression induite par les œstrogènes dans les cellules tumorales se trouve renforcée [40].
Ces exemples illustrent la difficulté de trancher entre des marques « causales » et des marques « conséquences » de longs processus physiopathologiques, auxquels s’ajoutent des perturbations environnementales exogènes et endogènes simultanées, liées au sexe et au vieillissement. Cette question, difficilement abordable expérimentalement chez l’homme, peut être abordée dans des modèles animaux appropriés.
Perspectives : quels enjeux pour l’épigénomique environnementale ?
L’augmentation de l’incidence des maladies chroniques à travers le monde représente une préoccupation majeure dans le domaine de la santé. Les nouvelles connaissances sur la DOHaD, en particulier les impacts de l’environnement sur la phase de programmation au cours du développement et ses mécanismes épigénétiques, représentent une opportunité pour combattre ce fléau notamment par la prévention. L’épigénétique contemporaine, moléculaire, est, elle, étudiée depuis plus de 40 ans, 30 ans dans le cadre du cancer. Il reste donc à combler le fossé entre les connaissances et les apports d’une recherche fondamentale de pointe sur l’épigénétique et les besoins d’une recherche appliquée dans le cadre de la DOHaD, à l’animal ou à l’humain. Il faudrait pour cela que l’on étudie les perturbations de l’environnement sur les processus épigénétiques, pendant toutes les phases y compris les phases précoces, ce qui est encore très peu abordé.
La complexité des processus épigénétiques et la variété des facteurs environnementaux nécessitent le développement d’une recherche transdisciplinaire, qui seule permettra d’imaginer de nouvelles approches, de découvrir des mécanismes encore insoupçonnés et de développer des stratégies de prévention adaptées, cohérentes et efficaces chez les sujets à risque.
Quant à la transmission aux générations suivantes de susceptibilité à des maladies chroniques ou d’une résilience, par le biais de mécanisme(s) épigénétique(s) qui sont encore mal étayés, elle n’est pas encore démontrée très clairement chez l’humain, mais les preuves s’accumulent chez le rongeur, les nématodes, la drosophile et surtout les plantes [54–58]. Jusqu’à présent un nombre important de revues portent sur les effets inter- ou transgénérationnels [15–17, 59]. Or cette phase, pour passionnante qu’elle soit, ne correspond qu’à l’une des trois grandes phases clés de la DOHaD. Si certains caractères sont effectivement transmissibles, le principal défi pour l’individu, et surtout pour les pouvoirs publics, est de pouvoir éviter ces transmissions.
Quoi qu’il en soit, ces données risquent de déplacer le curseur des responsabilités en termes de santé, de la sphère privée vers la sphère socio-géographico-politique et avec de nouvelles questions d’éthique. Cette revue souligne le rôle majeur de l’environnement sur notre santé. Un changement de paradigme s’impose pour passer du traitement, trop tardif, à la prévention. Pour cela une prise de conscience est nécessaire à tous les niveaux pour évaluer les véritables risques et prendre en compte les aspects coût/bénéfice des recommandations préconisées et validées. Compte tenu de l’impact des facteurs socioéconomiques on doit aussi, comme le fait P. O’Campo, s’interroger : « Apportons-nous les bonnes évidences/preuves scientifiques, qui permettront d’identifier les chaînons manquants et d’agir pour enrayer les effets des facteurs environnementaux et des déterminants sociaux de la santé ?» [60].
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
L’épisode Ice storm (tempête de pluie verglaçante) est une période de cinq jours consécutifs en janvier 1998 où une série de perturbations météorologiques provoquèrent des pluies verglaçantes dans l’est du Canada, la Nouvelle-Angleterre et le nord de l’État de New York. Le verglas a provoqué l’un des plus importants désastres naturels en Amérique du Nord.
Les attentats-suicides du 11 septembre 2001 ont été perpétrés aux États-Unis faisant 2 977 victimes.
The term “epigenetics” was coined by Conrad Hal Waddington in 1942 to mean the study of the “causal mechanisms” of development, which bring the phenotype into being.
In the context of molecular biology, epigenetics is defined as the study of mitotically or meiotically heritable changes in gene function that cannot be explained by changes in the DNA sequence.
The structural adaptation of chromosomal regions so as to register, signal or perpetuate altered activity states.
Les cellules ES (embryonic stem cells) sont artéfactuelles et ne sont qu’un modèle qui ne reproduit pas nécessairement ce qui se passe in vivo dans l’embryon précoce.
Références
- Eskenazi B, Marks AR, Catalano R, et al. Low birthweight in New York City and upstate New York following the events of September 11th. Hum Reprod 2007 ; 22 : 3013–3020. [CrossRef] [PubMed] [Google Scholar]
- Cao-Lei L, Massart R, Suderman MJ, et al. DNA methylation signatures triggered by prenatal maternal stress exposure to a natural disaster: project ice storm. PLoS One 2014 ; 9 : e107653. [CrossRef] [PubMed] [Google Scholar]
- Weaver ICG, Cervoni N, Champagne FA, et al. Epigenetic programming by maternal behavior. Nat Neurosci 2004 ; 7 : 847–854. [Google Scholar]
- McGowan PO, Suderman M, Sasaki A, et al. Broad epigenetic signature of maternal care in the brain of adult rats. PLoS One 2012 ; 6 : e14739. [CrossRef] [Google Scholar]
- Provencal N, Suderman MJ, Guillemin C, et al. Association of childhood chronic physical aggression with a DNA methylation signature in adult human T cells. PLoS One 2014 ; 9 : e89839. [CrossRef] [PubMed] [Google Scholar]
- Suderman M, McGowan PO, Sasaki A, et al. Conserved epigenetic sensitivity to early life experience in the rat and human hippocampus. Proc Natl Acad Sci USA 2012 ; 109 Suppl 2 : 17266–17272. [CrossRef] [Google Scholar]
- Gapp K, Jawaid A, Sarkies P, et al. Implication of sperm RNAs in transgenerational inheritance of the effects of early trauma in mice. Nat Neurosci 2014 ; 17 : 667–669. [CrossRef] [PubMed] [Google Scholar]
- McGowan PO, Sasaki A, D’Alessio AC, et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci 2009 ; 12 : 342–348. [CrossRef] [PubMed] [Google Scholar]
- Szyf M. Lamarck revisited: epigenetic inheritance of ancestral odor fear conditioning. Nat Neurosci 2014 ; 17 : 2–4. [CrossRef] [PubMed] [Google Scholar]
- Jordan B. Épigénétique et résilience. Med Sci (Paris) 2013 ; 29 : 325–328. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Klengel T, Mehta D, Anacker C, et al. Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat Neurosci 2013 ; 16 : 33–41. [CrossRef] [PubMed] [Google Scholar]
- Guillemin C, Provencal N, Suderman M, et al. DNA methylation signature of childhood chronic physical aggression in T cells of both men and women. PLoS One 2014 ; 9 : e86822. [CrossRef] [PubMed] [Google Scholar]
- Teh AL, Pan H, Chen L, et al. The effect of genotype and in utero environment on interindividual variation in neonate DNA methylomes. Genome Res 2014 ; 24 : 1064–1074. [CrossRef] [PubMed] [Google Scholar]
- Rando OJ, Verstrepen KJ. Timescales of genetic and epigenetic inheritance. Cell 2007 ; 128 : 655–668. [CrossRef] [PubMed] [Google Scholar]
- Meaney MJ, Ferguson-Smith AC. Epigenetic regulation of the neural transcriptome: the meaning of the marks. Nat Neurosci 2010 ; 13 : 1313–1318. [CrossRef] [PubMed] [Google Scholar]
- Daxinger L, Whitelaw E. Understanding transgenerational epigenetic inheritance via the gametes in mammals. Nat Rev Genet 2012 ; 13 : 153–162. [CrossRef] [PubMed] [Google Scholar]
- Lim JP, Brunet A. Bridging the transgenerational gap with epigenetic memory. Trends Genet 2013 ; 29 : 176–186. [CrossRef] [PubMed] [Google Scholar]
- Gueant JL, Daval JL, Vert P, Nicolas JP. Folates et programmation fœtale : rôle des mécanismes nutrigénomiques et épigénomiques. Bull Acad Natl Med 2012 ; 196 : 1829–1842. [PubMed] [Google Scholar]
- Allis CD, Berger SL, Cote J, et al. New nomenclature for chromatin-modifying enzymes. Cell 2007 ; 131 : 633–636. [Google Scholar]
- Waddington C. Canalisation of development and inheritance of acquired characters. Nature 1942 ; 152 : 563. [CrossRef] [Google Scholar]
- Riggs AD, Xiong Z. Methylation and epigenetic fidelity. Proc Natl Acad Sci U S A 2004 ; 101 : 4–5. [CrossRef] [PubMed] [Google Scholar]
- Bird A. Perceptions of epigenetics. Nature 2007 ; 447 : 396–398. [CrossRef] [PubMed] [Google Scholar]
- Orozco-Solis R, Sassone-Corsi P. Epigenetic control and the circadian clock: linking metabolism to neuronal responses. Neuroscience 2014 ; 264 : 76–87. [CrossRef] [PubMed] [Google Scholar]
- Rudenko A, Tsai LH. Epigenetic regulation in memory and cognitive disorders. Neuroscience 2014 ; 264 : 51–63. [CrossRef] [PubMed] [Google Scholar]
- Chi AS, Bernstein BE. Developmental biology. Pluripotent chromatin state. Science 2009 ; 323 : 220–221. [CrossRef] [PubMed] [Google Scholar]
- Graf T, Enver T. Forcing cells to change lineages. Nature 2009 ; 462 : 587–594. [CrossRef] [PubMed] [Google Scholar]
- Junien C, Gallou-Kabani C, Vige A, Gross MS. Épigénomique nutritionnelle du syndrome métabolique. Med Sci (Paris) 2005 ; 21 : 396–404. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Cech TR, Steitz JA. The noncoding RNA revolution-trashing old rules to forge new ones. Cell 2014 ; 157 : 77–94. [CrossRef] [PubMed] [Google Scholar]
- Ho SM, Johnson A, Tarapore P, et al. Environmental epigenetics and its implication on disease risk and health outcomes. ILAR J 2012 ; 53 : 289–305. [CrossRef] [PubMed] [Google Scholar]
- Junien C, Gabory A, Attig L. Le dimorphisme sexuel au XXIe siècle. Med Sci (Paris) 2012 ; 28 : 185–192. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- La Weber M. méthylation de l’ADN, un acteur-clé de la pluripotence. Med Sci (Paris) 2011 ; 27 : 483–485. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Ptashne M. Faddish stuff: epigenetics and the inheritance of acquired characteristics. FASEB J 2013 ; 27 : 1–2. [CrossRef] [PubMed] [Google Scholar]
- Branciamore S, Rodin AS, Riggs AD, Rodin SN. Enhanced evolution by stochastically variable modification of epigenetic marks in the early embryo. Proc Natl Acad Sci USA 2014 ; 111 : 6353–6358. [CrossRef] [Google Scholar]
- Ooi SK, O’Donnell AH, Bestor TH. Mammalian cytosine methylation at a glance. J Cell Sci 2009 ; 122 : 2787–2791. [CrossRef] [PubMed] [Google Scholar]
- Warner MJ, Ozanne SE. Mechanisms involved in the developmental programming of adulthood disease. Biochem J 2010 ; 427 : 333–347. [CrossRef] [MathSciNet] [PubMed] [Google Scholar]
- Gapp K, Woldemichael BT, Bohacek J, Mansuy IM. Epigenetic regulation in neurodevelopment and neurodegenerative diseases. Neuroscience 2014 ; 264 : 99–111. [CrossRef] [PubMed] [Google Scholar]
- Kelly-Irving M, Lepage B, Dedieu D, et al. Childhood adversity as a risk for cancer: findings from the 1958 British birth cohort study. BMC Public Health 2013 ; 13 : 767. [CrossRef] [PubMed] [Google Scholar]
- Heard E. The dynamics of epigenetic changes in a range of organisms. Curr Top Dev Biol 2013; 104 : xiii–xv. [CrossRef] [PubMed] [Google Scholar]
- Ozanne SE, Sandovici I, Constancia M. Maternal diet, aging and diabetes meet at a chromatin loop. Aging (Albany NY) 2011 ; 3 : 548–554. [CrossRef] [PubMed] [Google Scholar]
- Hsu PY, Hsu HK, Singer GA, et al. Estrogen-mediated epigenetic repression of large chromosomal regions through DNA looping. Genome Res 2010 ; 20 : 733–744. [CrossRef] [PubMed] [Google Scholar]
- Metivier R, Gallais R, Tiffoche C, et al. Cyclical DNA methylation of a transcriptionally active promoter. Nature 2008 ; 452 : 45–50. [CrossRef] [PubMed] [Google Scholar]
- Pinney SE, Simmons RA. Epigenetic mechanisms in the development of type 2 diabetes. Trends Endocrinol Metab 2010 ; 21 : 223–229. [CrossRef] [PubMed] [Google Scholar]
- Xue Z, Ye Q, Anson SR, et al. Transcriptional interference by antisense RNA is required for circadian clock function. Nature 2014 ; 514 : 650–653. [CrossRef] [PubMed] [Google Scholar]
- Woldemichael BT, Bohacek J, Gapp K, Mansuy IM. Epigenetics of memory and plasticity. Prog Mol Biol Transl Sci 2014 ; 122 : 305–340. [CrossRef] [PubMed] [Google Scholar]
- Mathias PC, Elmhiri G, de Oliveira JC, et al. Maternal diet, bioactive molecules, and exercising as reprogramming tools of metabolic programming. Eur J Nutr 2014 ; 53 : 711–722. [CrossRef] [PubMed] [Google Scholar]
- Weaver IC, Champagne FA, Brown SE, et al. Reversal of maternal programming of stress responses in adult offspring through methyl supplementation: altering epigenetic marking later in life. J Neurosci 2005 ; 25 : 11045–11054. [CrossRef] [PubMed] [Google Scholar]
- Burdge GC, Lillycrop KA, Phillips ES, et al. Folic acid supplementation during the juvenile-pubertal period in rats modifies the phenotype and epigenotype induced by prenatal nutrition. J Nutr 2009 ; 139 : 1054–1060. [CrossRef] [PubMed] [Google Scholar]
- Attig L, Vige A, Gabory A, et al. Dietary alleviation of maternal obesity and diabetes: increased resistance to diet-induced obesity transcriptional and epigenetic signatures. PLoS One 2013 ; 8 : e66816. [CrossRef] [PubMed] [Google Scholar]
- Remacle C, Dumortier O, Bol V, et al. Intrauterine programming of the endocrine pancreas. Diabetes Obes Metab 2007 ; 9 (suppl 2) : 196–209. [CrossRef] [PubMed] [Google Scholar]
- Edinger RS, Mambo E, Evans MI. Estrogen-dependent transcriptional activation and vitellogenin gene memory. Mol Endocrinol 1997 ; 11 : 1985–1993. [CrossRef] [PubMed] [Google Scholar]
- Pirola L, Balcerczyk A, Okabe J, El-Osta A. Epigenetic phenomena linked to diabetic complications. Nat Rev Endocrinol 2010 ; 6 : 665–675. [CrossRef] [PubMed] [Google Scholar]
- Miao F, Chen Z, Genuth S, et al. Evaluating the role of epigenetic histone modifications in the metabolic memory of type 1 diabetes. Diabetes 2014 ; 63 : 1748–1762. [CrossRef] [PubMed] [Google Scholar]
- Pirola L, Balcerczyk A, Tothill RW, et al. Genome-wide analysis distinguishes hyperglycemia regulated epigenetic signatures of primary vascular cells. Genome Res 2011 ; 21 : 1601–1615. [CrossRef] [PubMed] [Google Scholar]
- Grossniklaus U, Kelly WG, Ferguson-Smith AC, et al. Transgenerational epigenetic inheritance: how important is it? Nat Rev Genet 2013 ; 14 : 228–235. [CrossRef] [PubMed] [Google Scholar]
- Sela M, Kloog Y, Rechavi O. Non-coding RNAs as the bridge between epigenetic mechanisms, lineages and domains of life. J Physiol 2014 ; 592 : 2369–2373. [CrossRef] [PubMed] [Google Scholar]
- Cuzin F, Rassoulzadegan M. Non-Mendelian epigenetic heredity: gametic RNAs as epigenetic regulators and transgenerational signals. Essays Biochem 2010 ; 48 : 101–106. [CrossRef] [PubMed] [Google Scholar]
- Rechavi O, Houri-Ze’evi L, Anava S, et al. Starvation-induced transgenerational inheritance of small RNAs in C. elegans. Cell 2014 ; 158 : 277–287. [CrossRef] [PubMed] [Google Scholar]
- Heard E, Martienssen RA. Transgenerational epigenetic inheritance: myths and mechanisms. Cell 2014 ; 157 : 95–109. [CrossRef] [PubMed] [Google Scholar]
- Ferguson-Smith AC, Patti ME. You are what your dad ate. Cell Metab 2011 ; 13 : 115–117. [CrossRef] [PubMed] [Google Scholar]
- O’Campo P. Are we producing the right kind of actionable evidence for the social determinants of health? J Urban Health 2012 ; 89 : 881–893. [CrossRef] [PubMed] [Google Scholar]
- Lange UC, Schneider R. What an epigenome remembers. Bioessays 2010 ; 32 : 659–668. [CrossRef] [PubMed] [Google Scholar]
- Dardente H. Redondance génétique et synchronisation cellulaire dans les horloges circadiennes. Med Sci (Paris) 2008 ; 24 : 270–276. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
Liste des figures
|
Figure 1. Le cycle de la vie. La DOHaD comprend trois grandes phases : (1) la phase développementale de programmation de l’individu – couvrant la gestation, la petite enfance, et aussi l’adolescence – caractérisée par une grande plasticité de l’épigénome ; (2) la phase des effets à moyen et long terme, conséquences des pertur- bations développementales, mais auxquelles viennent s’associer, tout au long de la vie, les effets fluctuants ou permanents de l’environnement ; (3) la phase de reproduction puis les effets ou réponses inter- et trans- générationnels. Au cours de ces phases, les différentes perturbations subies par les marques épigénétiques sous l’influence de l’environnement modulent le phénotype. Bien que le terme « transgénérationnel » puisse être uti- lisé souvent indifféremment, on se doit de distinguer les effets transgénérationnels des effets intergénérationnels ou multigénérationnels. Les premiers impliquent une transmission par la lignée germinale, pour la première génération hors de tout contact avec l’agent environnemental perturbateur. Cet effet transgénérationnel ne peut apparaître qu’au niveau de la F3 par la lignée maternelle, ou de la F2 par la lignée paternelle. Les effets observés en deçà sont appelés intergénérationnels. Les effets multigénérationnels correspondent à une exposition au même stress ou à un stress qui peut découler de la première exposition, sur plusieurs généra- tions. Le point rouge représente, après la conception, le début de la gamétogenèse : le terme naissance reste postérieur. Enfin, il existe des différences entre les mâles et les femelles dues, dès la conception, à la différence entre les paires de chromosomes sexuels (XX et XY) puis, après la différenciation des gonades, à l’expression des hormones sexuelles. Des différences très précoces d’expression de gènes de la machinerie épigénétique apposent des marques épigénétiques spécifiques du sexe qui établissent de façon temporaire ou pérenne des réseaux fonctionnels de gènes différents chez le mâle et la femelle. La gamétogenèse et la résistance à la reprogrammation sont aussi différentes entre le mâle et la femelle. On observe ainsi un dimor- phisme sexuel, tant au niveau basal que pour les réponses à court terme ou à long terme [30]. |
| Dans le texte | |
|
Figure 2. Génétique et épigénétique. Notre patrimoine génétique composé de 3 milliards de paires de bases, identique dans toutes nos cellules, n’est pas nu dans le noyau : il entoure un complexe protéique formé de 4 paires d’histones formant une série de « perles » appelées nucléosomes. Des modifications réversibles appo- sées soit sur la séquence d’ADN (méthylation), soit sur les histones, participent au degré de compaction de la chromatine permettant ainsi à un gène de s’exprimer ou à l’inverse d’être réprimé. Ces mécanismes sont impliqués dans toute une série de mécanismes dont la transcription (expression), la réparation-répli- cation, la condensation, l’inactiva- tion de l’X, l’empreinte parentale, le vieillissement et le dimorphisme sexuel, etc. De plus, contrairement aux marques génétiques qui sont irréversibles, les marques épigénétiques, sensibles à tout type d’environnement, sont, par nature, réversibles. |
| Dans le texte | |
|
Figure 3. Héritabilité mitotique : de quoi la chromatine se souvient-elle et comment ? Mécanismes moléculaires sous-tendant la transmission épigénétique mitotique. Le schéma montre la réplication de l’ADN (bleu foncé, ADN parental ; bleu clair, ADN nouvellement synthétisé) à la fourche de réplication (flèche orange). Lors de la réplication, les nucléosomes parentaux (gris) sont dissociés et réassemblés sur les brins d’ADN néoformés en utilisant des histones parentales (gris) et nouvellement synthétisées (bleu). Les marques d’histone sont illustrées sous forme de carrés verts et les mécanismes responsables de leur transmission sont encadrées en vert. La méthylation de l’ADN est illustrée sous forme de cercles rouges et les mécanismes responsables de la main- tenance de la méthylation sont encadrées en rouge. La transmission mitotique des marques épigénétiques peut être dépendante de la réplication, par exemple via le couplage avec la fourche de réplication, ou indépendante de la machinerie de réplication. Actuellement, on connaît très peu de choses sur la façon dont les domaines de la chromatine active sont maintenus (tracé vert clair). Contrairement à la fidélité de recopiage de la séquence d’ADN et de la méthylation de l’ADN, la duplication des histones ne se fait pas à partir d’une matrice et les modifications post-traductionnelles ne sont pas conservées au moment où la cellule se divise. Les marques d’histones semblent s’apposer conformément à l’état transcriptionnel, mais après la réplication. Les gènes sont activés par des protéines régulatrices, les facteurs de transcription (FT), qui se fixent à l’ADN au niveau des promoteurs et enhancers. Au cours de la réplication cellulaire, ceux-ci sont distribués aux cellules filles qui les séquestrent car leur présence continuelle est requise. Les histones néosynthétisées viennent s’associer aux histones parentales porteuses des modifications post-traductionnelles, disloquées lors de la progression de la fourche de répli- cation. Des protéines de type chaperon les accompagnent. Contrairement à la méthylation de l’ADN, le mécanisme par lequel les profils de modifications post-traductionnelles des histones sont rétablis après chaque division demeure obscur et l’objet d’ardents débats. En effet, des altérations des systèmes Polycomb/trithorax sont souvent héritées par les générations suivantes de cellules et parfois d’organismes (d’après [61]). |
| Dans le texte | |
|
Figure 4. La mémoire est dynamique à travers l’es- pace, boucle, promoteurs, enhancers. Modèle simpli- fié proposé pour la boucle formée par l’association entre l’enhancer (Enh, activateur transcriptionnel) et le promoteur (P2) dans le locus Hnf4a (hepatocyte nuclear factor-4-alpha) dans les îlots pancréatiques d’animaux contrôles jeunes et âgés (rangée du haut) et dénutris en protéines (rangée du bas). Dans les îlots des jeunes rats adultes contrôles, l’enhancer (Enh) est enrichi en marques d’histone actives et interagit étroitement avec le promoteur P2 de Hnf4a, conduisant à des niveaux élevés de transcription de P2. L’interaction de l’enhancer-promoteur avec P2 est altérée dans les îlots des animaux exposés à une alimentation pauvre en protéines au cours du développement précoce. Ceci est dû à l’épuisement en marques d’histones activatrices et à l’enrichisse- ment en marques d’histones inhibitrices au niveau de la région de l’enhancer. Le vieillissement est associé à une réduction des niveaux d’expression d’Hnf4a en raison de l’enrichissement en marques d’histones inhibitrices le long du locus, et ceci est plus prononcé dans les îlots des animaux LP. L’accumulation de marques d’histones inhibitrices pourrait affaiblir davantage l’interaction de l’enhancer- promoteur avec P2. La différence entre les adultes contrôles et les adultes tests dans la figure se manifeste par un écartement de la base de la boucle entre le promoteur et l’enhancer, un écartement qui est déjà prononcé entre les jeunes adultes soumis à un régime normal par rapport aux adultes âgés. La différence entre les 2 régimes s’accroît chez les adultes âgés soumis à une exposition précoce au régime pauvre en protéine (d’après [39]). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.