")
")
| Issue |
Med Sci (Paris)
Volume 32, Number 1, Janvier 2016
Origine développementale de la santé et des maladies (DOHaD), environnement et épigénétique
|
|
|---|---|---|
| Page(s) | 15 - 20 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/20163201004 | |
| Published online | 5 février 2016 | |
Le concept des origines développementales de la santé
Évolution sur trois décennies
Developmental origin of health and adult diseases (DOHaD): evolution of a concept over three decades
1
Inserm, UMR1153, centre de recherche en épidémiologie et biostatistiques, Sorbonne Paris-Cité (CRESS), équipe de recherche sur les origines précoces de la santé et du développement de l’enfant; Paris Descartes université, 16, avenue Paul Vaillant Couturier, F-94807
Villejuif, France
2
UMR1027, université Toulouse III, équipe cancer et maladies chroniques : inégalités sociales de santé, accès primaire et secondaire aux soins, 37, allées Jules Guesde, 31069
Toulouse, France
3
Inserm, unité 1138, université Pierre et Marie Curie UMRS 1138 et mission Inserm associations, département de l’information scientifique et de la communication, 101, rue de Tolbiac, 75013
Paris, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Dans les années 1980, l’hypothèse d’une origine fœtale des maladies de l’adulte a été avancée par D. Barker et son équipe. Ce concept a ensuite évolué vers celui des origines développementales de la santé et des maladies. Les progrès de l’épidémiologie sociale, des neurosciences et de la toxicologie ont contribué à identifier les premières années de la vie comme une période clé pour la santé future. Enfin, l’épigénétique a apporté une crédibilité biologique au concept. La convergence de tous ces courants amène à conceptualiser la santé humaine dans un continuum complexe et dynamique de Lifecourse Health Development. De nombreux modèles animaux ont été développés pour tenter d’apporter des éléments décisifs de compréhension. Leurs contributions dans le domaine du diabète sont décrites dans cet article.
Abstract
In the 1980s, D. Barker and his team proposed the hypothesis of a fetal origin of adult diseases. The concept subsequently evolved into the developmental origins of health and diseases. Progresses in various domains such as social epidemiology, neuroscience, toxicology have contributed to establish the early years of life as a key period for future health. Finally, epigenetics has provided biological plausibility to long-term programming of health by early exposures. The convergence of all these currents has led to conceptualize human health in a complex and dynamic continuum, the Lifecourse Health Development, beginning in the prenatal period and covering the whole life. Many animal models have been developed to try to unravel the mechanisms involved. Their contributions are described in this paper with the example of type 2 diabetes.
© 2016 médecine/sciences – Inserm

Des origines fœtales des maladies aux origines développementales de la santé et des maladies de l’adulte
Dans les années 1980, des épidémiologistes anglais ont publié les premiers travaux indiquant que les régions de Grande-Bretagne dans lesquelles on observait une mortalité néonatale et infantile élevée dans les années 1920 étaient aussi celles dans lesquelles on constatait un taux important de mortalité d’origine cardiovasculaire dans les années 1970 [1, 2]. L’un d’entre eux, David Baker, a cherché à confirmer au niveau individuel et, cette fois-ci, dans un lieu géographique plus restreint, les relations potentielles entre des événements survenant aux deux extrémités de la vie. Il a reconstitué plusieurs cohortes « historiques » à partir des archives conservées minutieusement dans une ville (Preston) et un comté (Hertfordshire) d’Angleterre, et concernant les naissances survenues dans la première moitié du xxe siècle. Ces cohortes ont démontré une relation inverse entre le poids de naissance et la mortalité mais aussi diverses maladies chroniques (cardiovasculaire, bronchopneumopathie chronique, diabète de type 2, etc.), de même que la présence de facteurs de risque de ces maladies à l’âge l’adulte, chez les hommes comme chez les femmes [3].
Pour interpréter ces résultats, l’hypothèse d’une origine fœtale des maladies de l’adulte a été avancée. Elle supposait que la malnutrition maternelle ou fœtale (via un défaut de développement placentaire par exemple) à certaines périodes clés de la grossesse entraînait chez le fœtus des adaptations pour lui permettre de survivre in utero. Hales et Barker ont proposé qu’en réponse à une malnutrition maternelle, le fœtus développe un phénotype d’épargne (en anglais, thrifty phenotype) qui assure la survie de l’organisme en épargnant les organes vitaux comme le cerveau, aux dépens d’organes comme le foie, le pancréas, les reins et les muscles [4]. Ces adaptions, résultant en un poids de naissance diminué, entraîneraient par la suite une susceptibilité accrue aux maladies chroniques de l’adulte.
Cette hypothèse, et les travaux épidémiologiques sur lesquels elle reposait, ont été beaucoup critiqués initialement [5] sur des arguments méthodologiques : le nombre élevé de « perdus de vue » dans ces études reliant des événements aux deux extrémités de la vie, ou le faible statut économique de la famille dans l’enfance et à l’âge adulte qui suffisait à expliquer l’association observée. Cependant, de nombreuses autres cohortes ont confirmé ces relations entre faible poids de naissance et marqueurs de risque cardiovasculaire et ont permis de répondre, une à une, à chacune des critiques avancées. Parmi celles-ci, une étude comportant très peu de perdus de vue a permis de montrer que la relation persistait après prise en compte du niveau socioéconomique et de l’âge gestationnel [6]. Une autre a démontré que la relation poids de naissance-mortalité cardiovasculaire n’était pas expliquée par les facteurs de risque cardiovasculaire classiques à l’âge adulte [7]. De nouvelles preuves ont été apportées par le suivi de femmes dont la grossesse s’est déroulée pendant, juste avant ou juste après, la famine hollandaise induite par le blocus imposé par l’armée allemande en 1944 [8]. De plus, des études chez des sujets plus jeunes ont retrouvé le même type de relation entre faible poids de naissance et marqueurs de risque cardiovasculaire, indiquant que ces relations n’étaient pas uniquement le fait de conditions de vie du début du xxe siècle, mais qu’elles étaient toujours pertinentes pour des sujets nés après guerre dans des conditions plus favorables, y compris en France [9, 10].
Le faible poids à la naissance n’est pas la seule condition qui a été associée à un risque de pathologie à l’âge adulte. Des observations faites initialement chez des enfants de mères ayant un diabète de type 1, puis dans des populations à forte prévalence de diabète de type 2, comme les Indiens Pimas aux États-Unis, ont montré une incidence accrue d’obésité et d’intolérance au glucose associée à un poids de naissance élevé et au développement du fœtus dans un environnement intra-utérin hyperglycémique. En ce qui concerne le diabète de type 2, une relation en « U » avec le poids de naissance a été clairement démontrée, traduisant un risque augmenté de cette pathologie dans des conditions opposées : celles d’une croissance fœtale restreinte ou au contraire accélérée [11].
Beaucoup de ces travaux initiaux ont porté sur le poids de naissance. C’est en effet un indicateur facilement disponible, et correctement mesuré, de l’environnement intra-utérin. Son utilisation a beaucoup contribué aux fondements de la théorie initiale des origines fœtales des maladies.
Cependant, dès les années 1980, à la suite de premières études observationnelles montrant un meilleur développement cognitif, ou statut métabolique, chez les enfants allaités, des essais randomisés ont été initiés chez le nouveau-né prématuré comparant une préparation infantile enrichie en nutriments à une préparation standard pour nouveau-né à terme ou au lait maternel (de donneur). Ces essais ont clairement démontré un bénéfice sur le développement cognitif, évalué à 8 ans, des préparations enrichies en comparaison aux préparations standard, tout particulièrement chez les garçons [12]. Malgré un contenu énergétique inférieur, les prématurés ayant reçu du lait humain ont atteint un niveau de développement similaire aux prématurés ayant reçu des préparations enrichies. Ainsi, dans les conditions expérimentales de ces essais, l’administration d’une alimentation différente pendant quelques semaines après la naissance a eu des effets à long terme sur le développement de l’enfant : le terme de « programmation nutritionnelle précoce » a été employé pour la première fois pour décrire ces résultats.
Pour faire la synthèse de l’ensemble de ces observations, le concept des origines fœtales de la santé et des maladies s’est vite révélé trop limité et il a évolué vers celui des origines développementales. En effet, l’étude du développement et de l’évolution dans de nombreuses espèces vivantes, dont certains éléments sont décrits dans le paragraphe suivant, a bien mis en évidence l’importante plasticité du développement de nombreux organes et tissus au cours de cette période précoce, en réponse à des variations de l’environnement. Les observations rapportées plus haut sont donc apparues comme la confirmation de ce même phénomène chez l’homme [13]. La vitesse de croissance de l’organisme est encore très élevée chez l’homme dans les deux premières années de vie, elle ralentit ensuite : la période prénatale et les deux premières années de vie après la naissance sont donc des périodes de susceptibilité importante pour des effets de programmation à long terme. Cette notion a été popularisée par la terminologie des « 1 000 jours » entre le début de la grossesse et les deux ans de l’enfant.
La convergence avec d’autres domaines scientifiques
L’épidémiologie sociale, en explorant le rôle des déterminants sociaux pour expliquer les inégalités sociales de santé, a montré que les facteurs de risque connus pour les maladies chroniques courantes ne peuvent expliquer totalement le risque accru de maladies chroniques chez les sujets situés plus bas dans l’échelle sociale (gradient social de santé). Parmi les explications envisagées, l’idée selon laquelle les facteurs sociaux auraient un effet biologique direct qui soit susceptible d’expliquer une part du gradient social commence à être explorée. Cette hypothèse renvoie au concept d’incorporation biologique (embodiment) qui fait référence à la façon dont « l’environnement spécifique de chaque milieu social pénètre sous la peau » ou encore comment le milieu social devient biologique [14]. L’environnement socioéconomique, psychologique, influerait sur des processus biologiques susceptibles à leur tour d’influencer l’état de santé futur. L’effet de l’environnement serait d’autant plus important que l’exposition est précoce, à la faveur de la grande plasticité biologique – notamment cérébrale – observée à cette période. L’approche de type « épidémiologie biographique » (lifecourse analysis) offre un cadre théorique en épidémiologie pour tenter de mieux explorer ces liens. Cette approche combine à la fois le modèle de « périodes critiques », développé à partir des hypothèses de programmation biologique et de l’origine développementale des maladies, et le modèle de chaînes de causalité (pathways), qui postule qu’une exposition précoce survenant très tôt dans le développement d’un individu peut le placer sur une chaîne de risque, incluant des processus biologiques, des facteurs psychosociaux et comportementaux.
Le développement des neurosciences a bien mis en évidence la très grande plasticité du développement cérébral dans les premières années de la vie. Le rôle très important des interactions précoces entre le bébé et les adultes qui prennent soin de lui, en tout premier lieu ses parents, sur les fonctions neuroendocrines et l’établissement des réseaux neuronaux a été mis en lumière [15]. Des situations d’adversité dans l’enfance, incluant des carences affectives ou des violences, ont des conséquences à l’âge adulte, sur la santé mentale (dépression, troubles anxieux) mais aussi physique, incluant des liens avec les pathologies survenant à un âge avancé comme le cancer [16–18]. L’épidémiologie et les neurosciences convergent ainsi pour suggérer qu’une variété de situations et d’expositions peuvent entraîner des altérations dans la structure et l’organisation cérébrale et dans les mécanismes de réponse neuroendocriniens, immunologiques et du système nerveux autonome à des événements stressants [19] (→).
(→) Voir la Synthèse de M. Rincel et al., page 93 de ce numéro
En toxicologie, l’interférence de substances exogènes avec le développement normal aboutissant à une mort fœtale, à des malformations ou, à un degré moindre, à une diminution du poids de naissance, est connue et étudiée depuis longtemps. Mais, dans ce domaine également, se développe le concept de modifications plus modestes en réponse à des expositions environnementales à faibles doses, n’entraînant pas de conséquences détectables immédiatement, mais capables d’interférer avec les processus biologiques normaux et de causer des dysfonctionnements plus tard au cours de la vie. C’est le cas en particulier des substances à effet de perturbateurs endocriniens ou de toxiques affectant le développement cérébral [20] (→).
(→) Voir la Synthèse de C. Mauduit et al., page 45 de ce numéro
Enfin, l’épigénétique apporte une crédibilité biologique au concept de programmation à long terme par des expositions précoces [21]. En effet, elle permet de comprendre comment ces expositions peuvent s’incorporer biologiquement en agissant sur le niveau d’« expression » des gènes et modifier le déroulement du programme génétique en fonction de son environnement. Depuis une vingtaine d’années, des données scientifiques de plus en plus nombreuses ont mis en évidence le fait que des marques épigénétiques s’apposent sur les gènes tout au long du développement et, en particulier, au moment de la vie in utero (→).
(→) Voir la Synthèse de C. Junien et al., page 35 de ce numéro
Selon le stade du développement où elles sont apposées, elles sont susceptibles de modifier quantitativement le développement de certaines lignées cellulaires, ou simplement la machinerie cellulaire ou encore les interrelations physiques ou biologiques qui s’établissent entre cellules ou entre organes. Les organes ou systèmes affectés sont très dépendants du stade de développement concerné.
La convergence de tous ces courants, des plus biologiques, intégrant la biologie des systèmes, jusqu’à la psychologie et la sociologie, amène à conceptualiser la santé humaine dans un continuum complexe et dynamique de lifecourse health development (LCHD) débutant dès la période prénatale et couvrant la vie entière. Les conséquences sur et pour le système de santé commencent à être réfléchies [22] (Figure 1).
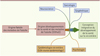 |
Figure 1. Évolution du concept des origines développementales de la santé (DOHaD). |
La preuve expérimentale par les modèles animaux
La croissance fœtale est un processus complexe et dynamique qui dépend étroitement de l’apport continu de nutriments par l’organisme maternel. Les différents organes se développent durant des fenêtres de croissance différentes. On comprend donc aisément combien une croissance inadéquate pendant ces périodes critiques peut « programmer » des défauts irréversibles de fonctionnement de l’organe, conduisant par la suite à des perturbations de l’équilibre physiologique.
Malgré les preuves épidémiologiques substantielles des origines fœtales des maladies de l’adulte, en particulier du diabète, les données obtenues chez l’homme à partir des cohortes longtemps rétrospectives et plus récemment prospectives, ont rendu possible l’étude des liens entre une exposition donnée et le risque de développer une pathologie sans toutefois permettre l’identification des mécanismes biologiques qui sous-tendent les processus physiopathologiques impliqués. C’est pourquoi de nombreux modèles animaux ont été développés pour tenter d’apporter des éléments décisifs de compréhension de ces processus. À l’heure actuelle, les espèces étudiées comprennent les rongeurs, les moutons et des primates non humains. Les traitements utilisés mettent en œuvre soit des restrictions alimentaires chez la femelle gestante (restriction protéique ou calorique globale), soit des restrictions vasculaires par ligature des artères utérines (provoquant une réduction de l’apport de nutriments au fœtus), soit l’induction d’un diabète ou d’une obésité expérimentale chez la mère (évoqués ultérieurement), soit un stress provoqué par la séparation maternelle précoce chez les rongeurs qui engendre des comportements anxieux ou dépressifs chez les descendants [23].
Dans la prochaine section, nous évoquerons principalement les modèles animaux dans le contexte du diabète ou du métabolisme, les hypothèses et les mécanismes qui en découlent, et comment les approches se sont complexifiées chez les rongeurs, en nous appuyant sur un exemple concret (Figure 2). Nous examinerons également un modèle de primates.
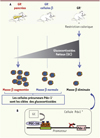 |
Figure 2. Restriction calorique au cours de la grossesse et développement du diabète de type 2 : apports des modèles de rongeurs. A. La restriction calorique au cours de la gestation entraîne une augmentation des glucocorticoïdes maternels et fœtaux qui à leur tour diminuent la masse de cellules bêta (à droite, de haut en bas). L’inactivation du récepteur des glucocorticoïdes (GR) dans les cellules précurseurs du pancréas par le système CreLoxP (souris GR- pancréas) a l’effet opposé et augmente le nombre de cellules bêta (à gauche) tandis que l’inactivation du GR dans les cellules bêta est sans effet (au centre). B. Le mécanisme (en bas) : les GC provoquent la fixation du GR sur le promoteur du gène Pdx-1 (pancreatic and duodenal homeobox 1) qui se lie avec un partenaire, PGC-1α (peroxisome proliferator-activated receptor gamma, coactivator 1 alpha). Cette liaison inhibe la transcription du gène Pdx-1, un gène clé du développement pancréatique, diminuant ainsi la masse de cellules bêta [25, 26]. GC : glucocorticoïdes ; GR : récepteur des glucocorticoïdes |
Modèles de restriction alimentaire
Selon l’hypothèse du phénotype d’épargne, proposée par Hales et Barker [4], le risque accru de développer un diabète dans des conditions de nutrition insuffisante in utero peut s’expliquer soit par un défaut de développement des cellules bêta pancréatiques au cours de la vie fœtale, à un moment où ces cellules se développent rapidement, soit par la constitution d’une insulinorésistance, soit encore, par l’association de ces deux composantes. En raison de la difficulté à vérifier cette hypothèse dans l’espèce humaine, des modèles animaux ont été créés. Les premiers ont été des modèles de restriction alimentaire réalisés chez la ratte gestante, en utilisant deux stratégies alimentaires différentes : la manipulation isocalorique faible en protéines ou la restriction nutritionnelle globale.
La restriction alimentaire protéique isocalorique : le modèle le plus ancien et le plus utilisé
Un régime isocalorique faible en protéines maternelles a été utilisé pour explorer les mécanismes par lesquels le métabolisme des protéines affecte le développement des organes. Snoeck et collègues ont démontré que la privation maternelle en protéines pendant toute la gestation réduit, chez les descendants, la masse de cellules bêta en diminuant la capacité de ces cellules à se multiplier [24]. Le maintien de cette malnutrition chez les descendants jusqu’à l’âge adulte conduit à une intolérance au glucose et une diminution de la réponse insulinique, alors qu’un retour à une nutrition normale, dès le sevrage, conduit à une situation intermédiaire, mais néanmoins altérée [25]. Ce modèle souligne que de brèves périodes de malnutrition, même suivies d’une nutrition adéquate, produisent des changements irréversibles sur le pancréas et son fonctionnement. Ce modèle a été utilisé pour répondre à de nombreuses questions sur les conséquences de la malnutrition sur le pancréas mais également sur le rein, et aussi pour explorer les maladies cardiovasculaires. Il est à noter que chez le rat, selon la composition protéique des régimes appauvris en protéines, les effets de programmation peuvent prédisposer la progéniture à différents effets physiopathologiques à l’âge adulte qui conduisent, ou non, à une hypertension artérielle [26].
La restriction alimentaire globale : un rôle important pour les glucocorticoïdes et le stress
La restriction de 50 % des apports nutritionnels maternels chez le rat, durant le dernier tiers de la gestation, provoque chez les fœtus un retard de croissance irréversible et une diminution de la masse de cellules bêta pancréatiques, qui persiste à l’âge adulte et conduit finalement à une intolérance au glucose [27].
Les états de dénutrition entraînent une activation de l’axe corticotrope et une sécrétion accrue de glucocorticoïdes (hypercorticisme) [28]. C’est le cas dans le modèle précité où il a été démontré que les hormones glucocorticoïdes jouaient un rôle prépondérant dans les anomalies observées [29, 30]. La Figure 2 montre comment l’utilisation de techniques sophistiquées, comme la génération de souris mutantes et l’immunoprécipitation de la chromatine, a permis de comprendre les mécanismes à l’origine du phénotype obtenu après restriction alimentaire, d’identifier la cellule cible – la cellule précurseur –, et finalement le gène cible, Pdx-1 (pancreatic and duodenal homebox 1), un facteur déterminant dans le développement des cellules bêta [29, 30].
Le modèle de restriction utéro-placentaire
Le modèle de restriction utéro-placentaire est un modèle expérimental chirurgical qui limite l’apport de substrats clés au fœtus : il se traduit par une diminution des taux de glucose, d’insuline, d’IGF-I (insulin growth factor), des acides aminés et de l’oxygène. Un tel modèle a été développé par l’équipe de Simmons chez le rat par ligature bilatérale des artères utérines trois jours avant la mise bas. Les animaux présentent un retard de croissance intra-utérin (RCIU) et développent progressivement un diabète et une insulinorésistance dès l’âge de 7 semaines. À cet âge, la masse des cellules bêta est normale. Elle s’épuise cependant progressivement pour ne représenter à l’âge de 6 mois qu’un tiers de la masse normale. Ce déficit est, en outre, associé à une obésité, un phénotype très similaire à celui observé chez l’homme atteint de diabète de type 2 [31].
Ce modèle est intéressant, car il a permis de démontrer un parallèle entre la diminution progressive de la masse de cellules bêta, celle du facteur de transcription Pdx-1 (pancreatic and duodenal homeobox 1) et les changements progressifs de la déacétylation des histones H3 et H4 au niveau du promoteur du gène codant Pdx-1. Ces changements épigénétiques précèdent le début du dysfonctionnement des cellules bêta suggérant leur rôle pathogénique dans ces modifications. Dans ce modèle, bien d’autres gènes jouant un rôle crucial dans la fonction et la prolifération des cellules bêta sont également la cible de modifications épigénétiques [32].
Réponse au stress et programmation du syndrome métabolique : des rongeurs aux primates
Un dysfonctionnement de la réponse au stress est soupçonné dans l’étiologie de plusieurs désordres métaboliques comme le diabète de type 2. Chez l’homme, il est clair que l’exposition prénatale à des taux élevés d’hormones glucocorticoïdes, par exemple lors de traitements de la mère aux corticostéroïdes comme la prednisone, ou dans le cas d’un syndrome de Cushing1, est associée à un retard de croissance intra-utérin [33], mais les conséquences métaboliques plus tardives ne sont pas connues. Nous avons vu qu’un stress élevé libère des hormones glucocorticoïdes [35] qui exercent sur le pancréas fœtal des rongeurs une empreinte qui a des conséquences à long terme. En effet, ces animaux, lorsqu’ils sont devenus adultes, présentent une intolérance au glucose.
Dans un modèle de primate à grossesse unique, le Chlorocebus aethiops ou singe vert, l’exposition prénatale à de faibles doses de dexaméthasone, un glucocorticoïde de synthèse, produit des troubles de l’homéostasie du glucose et de l’insuline, réduit la masse des cellules bêta pancréatiques, et augmente la pression artérielle et les niveaux de cortisol chez les descendants adolescents [34]. Ces données sont ainsi compatibles avec une extrapolation, faite sur les primates, de l’hypothèse de la programmation par les glucocorticoïdes. Elles suggèrent que la corticothérapie répétée et le stress chronique chez l’homme peuvent avoir des effets à long terme sur la régulation de l’homéostasie glucidique.
Conclusion
Tandis que la recherche épidémiologique a évolué pour montrer la contribution des origines développementales de la santé à de nombreuses maladies chroniques en les insérant dans des schémas physiopathologiques complexes au cours desquels interagissent et se succèdent plusieurs expositions délétères ou protectrices, la recherche chez l’animal s’est attachée à décrypter les mécanismes biologiques précis reliant une exposition précoce et ses effets à long terme, comme rappelé ici dans l’exemple de la malnutrition maternelle et de ses effets sur le pancréas de la descendance. D’autres exemples seront développés dans les différentes synthèses de ce numéro spécial. Faire converger ces approches parallèles mais complémentaires représente tout l’enjeu de la recherche sur l’origine développementale de la santé et des maladies de l’adulte (DOHaD) dans le futur. Les progrès techniques permettant d’appréhender, chez l’homme, la biologie systémique ouvrent de nouvelles possibilités. Mais les apports de ces premières décennies consacrées au concept DOHaD sont déjà suffisants pour tester l’effet d’un certain nombre d’interventions chez les mères et les jeunes enfants, par exemple dans le domaine nutritionnel, dont les résultats seront majeurs pour intégrer ce concept dans les stratégies de santé publique et thérapeutique.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Le syndrome de Cushing regroupe toutes les causes responsables d’un excès de sécrétion de cortisol par les glandes surrénales. Celles-ci incluent la maladie de Cushing – une tumeur bénigne de l’hypophyse qui produit trop d’hormone corticotrope (ACTH), ce qui entraîne une hypersécrétion de cortisol les 2 surrénales – ou des tumeurs d’autres organes, responsables d’une sécrétion ectopique d’ACTH.
Références
- Barker DJ, Osmond C. Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet 1986 ; 1 : 1077–1081. [CrossRef] [PubMed] [Google Scholar]
- Forsdahl A. Are poor living conditions in childhood and adolescence an important risk factor for arteriosclerotic heart disease? Br J Prev Soc Med 1977 ; 31 : 91–95. [PubMed] [Google Scholar]
- Barker DJP. Programming the baby. Mothers, babies and healt in later life. Edinburgh : Churchill Livingstone, 1998 : 13–42. [Google Scholar]
- Hales CN, Barker DJ. Type 2 (non-insulin-dependent) diabetes mellitus: the thrifty phenotype hypothesis. Diabetologia 1992 ; 35 : 595–601. [CrossRef] [PubMed] [Google Scholar]
- Joseph KS, Kramer MS. Review of the evidence on fetal and early childhood antecedents of adult chronic disease. Epidemiol Rev 1996 ; 18 : 158–174. [CrossRef] [PubMed] [Google Scholar]
- Leon DA, Lithell HO, Vagero D, et al. Reduced fetal growth rate and increased risk of death from ischaemic heart disease: cohort study of 15 000 Swedish men and women born 1915–29. BMJ 1998 ; 317 : 241–245. [CrossRef] [PubMed] [Google Scholar]
- Rich-Edwards JW, Stampfer MJ, Manson JE, et al. Birth weight and risk of cardiovascular disease in a cohort of women followed up since 1976. BMJ 1997 ; 315 : 396–400. [CrossRef] [PubMed] [Google Scholar]
- Roseboom T, de Rooij S, Painter R. The Dutch famine and its long-term consequences for adult health. Early Hum Dev 2006 ; 82 : 485–491. [CrossRef] [PubMed] [Google Scholar]
- Leger J, Levy-Marchal C, Bloch J, et al. Reduced final height and indications for insulin resistance in 20 year olds born small for gestational age: regional cohort study. BMJ 1997 ; 315 : 341–347. [CrossRef] [PubMed] [Google Scholar]
- Zureik M, Bonithon-Kopp C, Lecomte E, et al. Weights at birth and in early infancy, systolic pressure, and left ventricular structure in subjects aged 8 to 24 years. Hypertension 1996 ; 27 : 339–345. [CrossRef] [PubMed] [Google Scholar]
- McCance DR, Pettitt DJ, Hanson RL, et al. Birth weight and non-insulin dependent diabetes: thrifty genotype, thrifty phenotype, or surviving small baby genotype? BMJ 1994 ; 308 : 942–945. [CrossRef] [PubMed] [Google Scholar]
- Lucas A, Morley R, Isaacs E. Nutrition and mental development. Nutr Rev 2001 ; 59 : S24–S33. [CrossRef] [PubMed] [Google Scholar]
- Gluckman PD, Hanson MA, Bateson P, et al. Towards a new developmental synthesis: adaptive developmental plasticity and human disease. Lancet 2009 ; 373 : 1654–1657. [CrossRef] [PubMed] [Google Scholar]
- Krieger N. Embodiment: a conceptual glossary for epidemiology. J Epidemiol Community Health 2005 ; 59 : 350–355. [CrossRef] [PubMed] [Google Scholar]
- Cirulli F, Berry A, Alleva E. Early disruption of the mother-infant relationship: effects on brain plasticity and implications for psychopathology. Neurosci Biobehav Rev 2003 ; 27 : 73–82. [CrossRef] [PubMed] [Google Scholar]
- Horwitz AV, Widom CS, McLaughlin J, et al. The impact of childhood abuse and neglect on adult mental health: a prospective study. J Health Soc Behav 2001 ; 42 : 184–201. [CrossRef] [PubMed] [Google Scholar]
- Caspi A, Harrington H, Moffitt TE, et al. Socially isolated children 20 years later: risk of cardiovascular disease. Arch Pediatr Adolesc Med 2006 ; 160 : 805–811. [CrossRef] [PubMed] [Google Scholar]
- Kelly-Irving M, Lepage B, Dedieu D, et al. Childhood adversity as a risk for cancer: findings from the 1958 British birth cohort study. BMC Public Health 2013 ; 13 : 767. [CrossRef] [PubMed] [Google Scholar]
- Teicher MH, Andersen SL, Polcari A, et al. Developmental neurobiology of childhood stress and trauma. Psychiatr Clin North Am 2002 ; 25 : 397–426. [CrossRef] [PubMed] [Google Scholar]
- Barouki R, Gluckman PD, Grandjean P, et al. Developmental origins of non-communicable disease: implications for research and public health. Environ Health 2012 ; 11 : 42. [CrossRef] [PubMed] [Google Scholar]
- Junien C. Les déterminants précoces de la santé et des maladies : épigénéique et environnement. Bull Acad Nat Med 2011 ; 195 : 511–527. [Google Scholar]
- Halfon N, Larson K, Lu M, et al. Lifecourse health development: past, present and future. Matern Child Health J 2014 ; 18 : 344–365. [CrossRef] [PubMed] [Google Scholar]
- Uchida S, Hara K, Kobayashi A, et al. Early life stress enhances behavioral vulnerability to stress through the activation of REST4-mediated gene transcription in the medial prefrontal cortex of rodents. J Neurosci 2010 ; 30 : 15007–15018. [CrossRef] [PubMed] [Google Scholar]
- Snoeck A, Remacle C, Reusens B, et al. Effect of a low protein diet during pregnancy on the fetal rat endocrine pancreas. Biol Neonate 1990 ; 57 : 107–118. [CrossRef] [PubMed] [Google Scholar]
- Dahri S, Snoeck A, Reusens-Billen B, et al. Islet function in offspring of mothers on low-protein diet during gestation. Diabetes 1991 ; 40 : (suppl 2) : 115–120. [CrossRef] [PubMed] [Google Scholar]
- Langley-Evans SC. Critical differences between two low protein diet protocols in the programming of hypertension in the rat. Int J Food Sci Nutr 2000 ; 51 : 11–17. [CrossRef] [PubMed] [Google Scholar]
- Garofano A, Czernichow P, Breant B. Effect of ageing on beta-cell mass and function in rats malnourished during the perinatal period. Diabetologia 1999 ; 42 : 711–718. [CrossRef] [PubMed] [Google Scholar]
- Nyirenda MJ, Seckl JR. Intrauterine events and the programming of adulthood disease: the role of fetal glucocorticoid exposure. Int J Mol Med 1998 ; 2 : 607–614. [PubMed] [Google Scholar]
- Breant B, Gesina E, Blondeau B. Nutrition, glucocorticoids and pancreas development. Horm Res 2006 ; 65 : (suppl 3) : 98–104. [CrossRef] [PubMed] [Google Scholar]
- Valtat B, Riveline JP, Zhang P, et al. Fetal PGC-1alpha overexpression programs adult pancreatic beta-cell dysfunction. Diabetes 2013 ; 62 : 1206–1216. [CrossRef] [PubMed] [Google Scholar]
- Simmons RA, Templeton LJ, Gertz SJ. Intrauterine growth retardation leads to the development of type 2 diabetes in the rat. Diabetes 2001 ; 50 : 2279–2286. [CrossRef] [PubMed] [Google Scholar]
- Pinney SE, Jaeckle Santos LJ, Han Y, et al. Exendin-4 increases histone acetylase activity and reverses epigenetic modifications that silence Pdx1 in the intrauterine growth retarded rat. Diabetologia 2011 ; 54 : 2606–2614. [CrossRef] [PubMed] [Google Scholar]
- Reinisch JM, Simon NG, Karow WG, et al. Prenatal exposure to prednisone in humans and animals retards intrauterine growth. Science 1978 ; 202 : 436–438. [CrossRef] [PubMed] [Google Scholar]
- De Vries A, Holmes MC, Heijnis A, et al. Prenatal dexamethasone exposure induces changes in nonhuman primate offspring cardiometabolic and hypothalamic-pituitary-adrenal axis function. J Clin Invest 2007 ; 117 : 1058–1067. [CrossRef] [PubMed] [Google Scholar]
- Moisan MP, Le Moal M. Le stress dans tous ses états. Med Sci (Paris) 2012 ; 28 : 612–617. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
Liste des figures
|
Figure 1. Évolution du concept des origines développementales de la santé (DOHaD). |
| Dans le texte | |
|
Figure 2. Restriction calorique au cours de la grossesse et développement du diabète de type 2 : apports des modèles de rongeurs. A. La restriction calorique au cours de la gestation entraîne une augmentation des glucocorticoïdes maternels et fœtaux qui à leur tour diminuent la masse de cellules bêta (à droite, de haut en bas). L’inactivation du récepteur des glucocorticoïdes (GR) dans les cellules précurseurs du pancréas par le système CreLoxP (souris GR- pancréas) a l’effet opposé et augmente le nombre de cellules bêta (à gauche) tandis que l’inactivation du GR dans les cellules bêta est sans effet (au centre). B. Le mécanisme (en bas) : les GC provoquent la fixation du GR sur le promoteur du gène Pdx-1 (pancreatic and duodenal homeobox 1) qui se lie avec un partenaire, PGC-1α (peroxisome proliferator-activated receptor gamma, coactivator 1 alpha). Cette liaison inhibe la transcription du gène Pdx-1, un gène clé du développement pancréatique, diminuant ainsi la masse de cellules bêta [25, 26]. GC : glucocorticoïdes ; GR : récepteur des glucocorticoïdes |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.