")
")
| Issue |
Med Sci (Paris)
Volume 31, Number 5, Mai 2015
|
|
|---|---|---|
| Page(s) | 529 - 537 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/20153105015 | |
| Published online | 9 juin 2015 | |
La conception de vecteurs adaptés à la thérapie génique oculaire
Developments in gene delivery vectors for ocular gene therapy
1
Inserm UMR S968, Institut de la vision, 17, rue Moreau, 75012
Paris, France
2
Sorbonne universités, UPMC université Paris 6, UMR S968, 75012
Paris, France
3
CNRS, UMR 7210, 75012
Paris, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
L’utilisation d’un gène comme médicament est l’un des concepts les plus passionnants de la médecine moderne. Les vecteurs viraux ont été largement utilisés à cette fin et ont montré une efficacité thérapeutique dans une variété de modèles animaux de dégénérescence rétinienne. L’extension de ce succès à une application clinique a été initialement lente, mais une expression à long terme de gènes thérapeutiques a récemment été obtenue chez des patients atteints de déficits immunitaires, d’hémophilie B ou de troubles héréditaires de la rétine. Ces résultats ont suscité des espoirs pour le traitement de nombreuses autres maladies, et ont ouvert la voie au développement de nouveaux outils de transfert de gènes. Comme nous le verrons ici, les perspectives et les défis de la thérapie génique sont dans une large mesure dépendants du tissu ciblé, de la maladie concernée et, surtout, de l’efficacité du transfert génique. L’acheminement du gène vers les cellules cibles dépend de vecteurs, qui doivent assurer une expression durable du gène sans engendrer ni toxicité ni réaction immunitaire de la part de l’hôte. La conception de tels vecteurs, proposée pour la première fois dans les années 1970, s’est avérée être plus compliquée que prévu, limitant pendant de nombreuses années le succès de la thérapie génique. Les vecteurs doivent donc être adaptés à chaque scénario ; nous discuterons ici la conception de tels vecteurs pour un transfert de gènes vers la rétine.
Abstract
Gene therapy is quickly becoming a reality applicable in the clinic for inherited retinal diseases. Its remarkable success in safety and efficacy, in clinical trials for Leber’s congenital amaurosis (LCA) type II generated significant interest and opened up possibilities for a new era of retinal gene therapies. Success in these clinical trials was mainly due to the favorable characteristics of the retina as a target organ. The eye offers several advantages as it is readily accessible and has some degree of immune privilege making it suitable for application of viral vectors. The viral vectors most frequently used for retinal gene delivery are lentivirus, adenovirus and adeno-associated virus (AAV). Here we will discuss the use of these viral vectors in retinal gene delivery with a strong focus on favorable properties of AAV. Thanks to its small size, AAV diffuses well in the inter-neural matrix making it suitable for applications in neural retina. Building on this initial clinical success with LCA II, we have now many opportunities to extend this proof-of-concept to other retinal diseases using AAV as a vector. This article will discuss what are some of the most imminent cellular targets for such therapies and the AAV toolkit that has been built to target these cells successfully. We will also discuss some of the challenges that we face in translating AAV-based gene therapies to the clinic.
© 2015 médecine/sciences – Inserm
La vue est un de nos sens les plus précieux et la cécité est donc un déficit sensoriel très handicapant. La rétine, située au fond de l’œil, est composée de différents types cellulaires, dont cinq grands types neuronaux, traitant l’information lumineuse. La rétine est le siège de nombreuses pathologies entraînant une cécité. La lumière est captée par un neurone particulier appelé le photorécepteur (PR). Une grande partie des maladies cécitantes, comme par exemple la dégénérescence maculaire liée à l’âge (DMLA) ou la rétinopathie pigmentaire, sont dues à la dégénérescence des photorécepteurs. Les dégénérescences rétiniennes héréditaires sont hétérogènes et complexes en raison de la grande diversité de leurs causes génétiques ; on recense à ce jour plus de 180 locus impliqués dans les rétinopathies liées à la mort cellulaire des photorécepteurs [1]. Celle-ci peut être la conséquence d’un défaut primaire des photorécepteurs ou être secondaire à un défaut de fonctionnement de la couche cellulaire sous-jacente aux photorécepteurs, appelée l’épithélium pigmentaire rétinien (EPR). Il n’y a actuellement pas de traitement médicamenteux permettant de cibler ces maladies à long terme.
La thérapie génique, qui vise à transférer un gène thérapeutique dans les cellules cibles afin qu’elles produisent elles-mêmes le « médicament » de façon stable et prolongée, est donc une stratégie thérapeutique particulièrement intéressante dans le cas des maladies de la rétine. Pour ce faire, des vecteurs ont été développés afin de transporter les gènes thérapeutiques d’intérêt et les délivrer dans les cellules cibles. Les récents succès de la thérapie génique oculaire pour le traitement d’une dégénérescence rétinienne appelée l’amaurose congénitale de Leber 2–4, dans trois essais cliniques indépendants en 2008, ont fait naître l’espoir que cette stratégie puisse être appliquée à d’autres maladies cécitantes [5].
Actuellement, la recherche dans ce domaine a pour objectifs de comprendre la physiopathologie de ces atteintes de la rétine, d’identifier des gènes thérapeutiques pertinents et plus efficaces pour le traitement de telles maladies, mais également de mettre au point des vecteurs plus performants pour pouvoir cibler la rétine et y transférer ces gènes thérapeutiques.
L’œil, un organe cible idéal pour la thérapie génique
L’œil présente des caractéristiques qui en font un organe cible idéal pour la thérapie génique. Premièrement, il est facile à atteindre par voie chirurgicale (Figure 1). Pour accéder à la rétine, située sur la face postérieure du globe oculaire, il existe deux voies d’administration : la voie intra-vitréenne et la voie sous-rétinienne. De plus, l’œil se prête bien aux traitements locaux, permettant ainsi d’obtenir des concentrations locales élevées de vecteurs. La barrière hémato-rétinienne séparant le milieu rétinien de la circulation sanguine, les vecteurs administrés restent ainsi piégés sans causer d’effets systémiques indésirables. Par ailleurs, l’œil bénéficie d’un privilège immun, ce qui est très avantageux pour l’application de vecteurs viraux. Enfin, les cellules rétiniennes sont postmitotiques, tout transfert de gène que nous effectuons vers ces cellules est donc en principe permanent. Ce sont ces aspects de l’organe visuel qui ont permis à la thérapie génique oculaire de progresser en première ligne des essais cliniques et d’obtenir le premier succès clinique dans ce domaine [6].
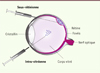 |
Figure 1. L’anatomie de l’œil humain et les voies d’injection intra-oculaires. L’œil humain focalise la lumière sur la rétine (en rose) à l’aide du cristallin, notamment sur la fovéa, la structure responsable de la vision à haute acuité. L’information lumineuse reçue est convertie en signaux électriques, puis transmise au cerveau par le nerf optique. L’injection sous-rétinienne consiste à déposer le vecteur dans l’espace sous-rétinien, entre la rétine et l’épithélium sous-jacent (EPR), directement en contact avec la rétine. Cette voie d’administration entraîne un décollement transitoire de la rétine qui se résorbe par la suite. L’injection intra-vitréenne amène, quant à elle, le vecteur dans le corps vitré, où il va ensuite diffuser et atteindre la rétine. |
Les cellules cibles de la thérapie génique rétinienne
La rétine est composée de trois couches de neurones, de cellules gliales et d’une couche d’épithélium pigmentaire (Figure 2).
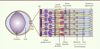 |
Figure 2. Structure de l’œil et de la rétine de souris. L’œil de souris est constitué d’un large cristallin et d’un petit volume de vitré (plus aqueux que le vitré de l’œil humain). La rétine est composée de plusieurs types neuronaux : les neurones de premier ordre sont les photorécepteurs (les cônes et les bâtonnets) ; les neurones de second ordre sont les cellules bipolaires, amacrines et horizontales ; et les cellules ganglionnaires sont les neurones de troisième ordre. L’épithélium pigmentaire (EPR) se situe derrière la rétine neurale, en contact avec les photorécepteurs. Les cellules gliales de Müller traversent verticalement la rétine, leurs pieds étant, d’une part, apposés au corps vitré et secrétant un réseau dense de polysaccharides (formant ainsi la membrane limitante interne [MLI]) et, d’autre part, apposés aux segments internes des photorécepteurs. |
La capture de lumière se fait au niveau des photorécepteurs, les neurones primaires de la rétine. Ils convertissent l’information lumineuse en signaux électriques qu’ils transmettent aux neurones de second ordre (cellules bipolaires, amacrines et horizontales) qui, à leur tour, les transmettent aux cellules ganglionnaires. Les informations transmises par ces neurones de troisième ordre sont communiquées au cerveau via le nerf optique. Le cerveau interprète alors les signaux transmis par la rétine.
L’épithélium rétinien pigmentaire est une couche monocellulaire pigmentée enveloppant les segments externes des photorécepteurs, essentiels à la vision et dans lesquels sont concentrés les pigments visuels. Lors du passage de la lumière, les segments externes sont dégradés et forment alors des résidus phagocytés par l’épithélium rétinien pigmentaire ; il y a une régénération permanente des segments externes. L’épithélium rétinien pigmentaire contient également des enzymes impliqués dans le recyclage des rétinoïdes pendant le cycle visuel qui a lieu dans les photorécepteurs et l’épithélium rétinien pigmentaire.
La rétine est également composée de trois types de cellules gliales permettant le maintien de l’homéostasie rétinienne : la microglie, l’astrocyte et la cellule gliale de Müller, cette dernière étant le type majoritaire. Les cellules gliales de Müller forment un soutien structural au tissu rétinien, assurant également des fonctions métaboliques et neuroprotectrices indispensables aux neurones de la rétine.
Les maladies génétiques conduisant à une cécité sont nombreuses et diverses, mais une grande partie d’entre elles sont dues à des mutations dans des gènes exprimés par l’épithélium rétinien pigmentaire ou les photorécepteurs. Ces cellules sont donc les cibles primaires des thérapies géniques visant à remplacer des gènes à l’origine de pathologies rétiniennes héréditaires.
Les principaux vecteurs utilisés pour un transfert de gène vers la rétine
Un vecteur permet de transporter du matériel génétique, par exemple un gène thérapeutique, à l’intérieur de cellules cibles. Deux grandes catégories de vecteurs sont utilisées pour cibler les cellules rétiniennes : les vecteurs synthétiques [7], qui sont issus d’une synthèse chimique, et les vecteurs viraux [8], qui sont obtenus à partir de virus génétiquement modifiés.
Les vecteurs viraux ont l’avantage d’être fondés sur un système de transfert de gène très efficace [9], celui des virus, qui ont évolué au cours de milliers d’années pour perfectionner leur art de véhiculer les acides nucléiques vers le noyau des cellules de mammifères. L’inconvénient de ces vecteurs est lié à la coévolution de leurs hôtes et, donc, des mécanismes de défense de ces derniers contre les infections virales. Les vecteurs synthétiques, tels que les composés lipidiques ou les polymères, ainsi que les peptides et les composés capables de compacter l’ADN en nanoparticules, sont les plus utilisés parmi les vecteurs synthétiques. Ils ont plusieurs avantages, notamment leur facilité de production (en quantités importantes) et leur capacité à transporter des séquences d’ADN de grande taille. Contrairement aux virus, ils présentent peu de risques de pathogénicité et leur utilisation in vitro donne de très bons résultats. En revanche, leur efficacité in vivo reste faible.
Nous décrirons ici l’utilisation des vecteurs viraux de transfert génique vers une rétine de rongeur, qui représente l’application la plus répandue en recherche aujourd’hui. Nous discuterons également les problèmes liés à la translation de ces résultats vers des modèles animaux précliniques, tels que le primate non humain.
Les vecteurs viraux sont les vecteurs les plus utilisés pour la thérapie génique in vivo. Les lentivirus, adénovirus et virus adéno-associés (AAV) (Tableau I) sont principalement étudiés pour le transfert génique vers la rétine des mammifères. Néanmoins, certains de ces virus ont des propriétés immunogènes, et peuvent aussi présenter un risque d’insertion nocive dans les chromosomes de la cellule hôte (interférant alors avec l’expression génique de l’hôte), ce qui représente une barrière supplémentaire à leur utilisation pour une application thérapeutique.
Caractéristiques des trois principaux vecteurs viraux utilisés en thérapie génique oculaire. kb : kilobases.
Historiquement, il a été supposé que l’expression d’un transgène à long terme nécessite son intégration dans le génome nucléaire, mais ce n’est pas le cas pour les cellules postmitotiques. Or, la rétine est généralement considérée comme étant composée entièrement de cellules postmitotiques. Ainsi, la perte par dilution d’un transgène épisomal en raison de la division cellulaire dans un tissu mitotique n’est pas un obstacle important pour le transfert de gènes vers la rétine.
Les adénovirus
Le tropisme d’un vecteur viral pour la rétine, et donc sa capacité à transduire les cellules y résidant, dépendent du stade de développement étudié. En effet, lors d’une administration sous-rétinienne chez la souris adulte, l’adénovirus (Ad) transduit essentiellement l’épithélium rétinien pigmentaire, en dépit de sa capacité à transduire en plus des photorécepteurs dans la rétine néonatale [10]. D’autre part, avec l’adénovirus, l’expression du transgène est rapide, mais de courte durée [10, 11]. Ceci est dû à l’élimination - par les lymphocytes T - des cellules hôtes transduites qui expriment les protéines de l’adénovirus codées par le génome viral [11]. Malgré ces limitations, plusieurs études ont indiqué que le transfert de gènes avec ce type de virus conduit à des améliorations de la vision à court terme dans des modèles animaux de dégénérescence rétinienne [12]. Dans ces études, l’adénovirus a été administré avant le 10e jour postnatal, donc avant que son tropisme pour les photorécepteurs et l’épithélium rétinien pigmentaire ne soit restreint à ce dernier.
Plus récemment, des adénovirus dépendants d’auxiliaires, qui ne codent pas pour des protéines virales [13] et qui échappent à la réponse immunitaire, ont été développés. Ces derniers ciblent l’épithélium rétinien pigmentaire de façon stable. Le développement des adénovirus, capables de transduire efficacement et de façon stable la rétine, serait particulièrement utile pour traiter les formes de dégénérescence rétinienne causées par des mutations dans des gènes de grande taille, trop larges pour être véhiculés par des vecteurs issus de virus adéno-associés ou de lentivirus, mais compatibles avec les adénovirus, en raison de la capacité élevée de chargement d’ADN (jusqu’à 36 kb) de ces derniers (Tableau I). De tels vecteurs permettraient aussi d’étudier les grandes régions de régulation de l’expression génique (par exemple, des amplificateurs endogènes [enhancers] ou des promoteurs) qui assureraient une régulation plus physiologique de l’expression d’un transgène. Ces vecteurs sont intéressants pour cibler des maladies dues à des mutations dans l’épithélium rétinien pigmentaire ; néanmoins, ils restent peu utilisés en raison des difficultés inhérentes à leur méthode de production.
Les lentivirus
Les lentivirus (Lv) sont des rétrovirus à ARN ayant une capacité de chargement d’ADN d’environ 8 à 10 kb ; ils s’intègrent dans le génome de l’hôte [14]. Bien qu’aucune transformation maligne n’ait été rapportée dans les yeux de souris en utilisant des titres élevés de lentivirus [14], la question de la mutagenèse insertionnelle continue d’être étudiée par la génération de lentivirus dits « non intégrants » et l’utilisation de lentivirus issus de primates non humains [14].
De nombreuses études montrent que, lorsqu’ils sont injectés par voie sous-rétinienne, les lentivirus transduisent efficacement l’épithélium rétinien pigmentaire chez les rongeurs [15, 16], mais la transduction des photorécepteurs reste variable [17, 18]. L’efficacité de transduction des photorécepteurs par les lentivirus dépend de l’origine du lentivirus et de l’espèce étudiée [19]. Par exemple, les lentivirus d’origine équine donnent de bons résultats de transduction des photorécepteurs situés à proximité du site d’injection chez le primate non humain [19, 20]. Deux essais cliniques sont en cours pour traiter la DMLA (NCT01301443), la maladie de Stargardt1 (STGD) (NCT01367444), et le syndrome d’Usher de type IB2 (USH1B) (NCT01505062), qui utilisent des lentivirus d’origine équine. Les résultats de ces essais ne sont pas encore rendus publics.
Les virus adéno-associés (AAV)
Biologie de l’AAV et production de vecteurs AAV recombinants
Les virus adéno-associés font partie de la famille des Parvoviridae, des virus non enveloppés dont le génome est formé d’ADN simple brin. Ces virus infectent la rétine neurale ainsi que l’épithélium rétinien pigmentaire, indépendamment de l’âge chez le rongeur [21, 22], et ils ont été utilisés avec succès chez l’homme [23, 24]. Les virus adéno-associés de type sauvage ne peuvent se répliquer qu’en présence d’un virus auxiliaire, comme par exemple l’adénovirus, le virus de l’herpès, ou le cytomégalovirus. À ce jour, plus d’une centaine de variants génétiques de virus adéno-associés naturels ont été isolés, mais seule une fraction d’entre eux sont caractérisés (Tableau II).
Récepteurs cellulaires de différents sérotypes d’AAV 25–27. EGFR : epidermal growth factor receptor ; FGF : fibroblast growth factor ; PDGF : platelet-derived growth factor.
La capside des virus adéno-associés forme un icosaèdre d’environ 25 nm de diamètre. Elle est constituée des protéines structurales VP1 (viral protein 1), VP2 et VP3, assemblées selon un ratio de 1:1:10. Le tropisme des variants d’AAV est déterminé principalement par les domaines en boucles des protéines VP. Les récepteurs cellulaires des virus adéno-associés ont été partiellement identifiés (Tableau II) et sont, pour certains, exprimés de façon ubiquitaire en surface des cellules mammifères.
Les mécanismes d’entrée des virus adéno-associés dans les cellules cibles diffèrent en fonction du sérotype [28]. De manière générale, l’infection par un virus adéno-associé commence par une adhésion aux récepteurs cellulaires suivie d’une internalisation par endocytose à travers des récepteurs secondaires. Suite à son transport endosomal et cytoplasmique, le virus est ensuite décapsidé et libère son ADN à l’intérieur du noyau, où il peut y avoir, soit une intégration spécifique dans le chromosome 19 (pour le virus sauvage), soit une concatémérisation3 épisomale (pour le virus recombinant).
Les virus adéno-associés recombinants constituent des vecteurs de choix, car ils permettent une expression forte et stable du transgène dans la rétine tout en étant faiblement immunogènes, et n’induisent pas d’effet cytopathique ou cancérigène. L’efficacité du transfert de gènes dans la rétine, comme dans d’autres tissus, dépend essentiellement de : (1) la capacité du protocole d’administration du vecteur adéno-associé à distribuer celui-ci dans toute la rétine, et le mettre ainsi en contact avec toutes les cellules à traiter ; (2) la capacité du vecteur à entrer dans la cellule et à transporter le transgène dans le noyau ; et (3) la capacité du transgène à être exprimé de façon forte et stable par les cellules transduites.
Le sérotype 2 du virus adéno-associé (AAV2) est le sérotype le plus étudié et le mieux caractérisé. C’est un excellent vecteur de transfert de gènes pour la transduction de certaines cellules, comme les cellules de l’épithélium rétinien pigmentaire ou les cellules ganglionnaires de la rétine. Cependant, il n’est pas efficace pour la transduction de nombreux autres types cellulaires, comme par exemple les photorécepteurs. De plus, la transduction par ce vecteur peut potentiellement être bloquée par les anticorps neutralisants dirigés contre des épitopes de la capside virale de l’AAV2. Afin de mieux cibler un tissu ou un type cellulaire d’intérêt, il sera judicieux de choisir une capside alternative à celle de l’AAV2. En effet, les différences de composition, et donc de structure de la capside, entre chaque sérotype de virus adéno-associé définissent le tropisme tissulaire et cellulaire de chacun d’entre eux. La production de vecteurs adéno-associés est facilitée par la génétique de ce virus : il est en effet possible d’encapsider des génomes à partir de celui de l’AAV2, en conservant l’ORF rep (open reading frame rep) impliquée dans la réplication du vecteur, et en y remplaçant la séquence codant pour la capside d’AAV2 par une séquence codant pour une capside alternative (Figure 3).
 |
Figure 3. Production de vecteurs AAV recombinants (AAVr) à partir d’AAV2. Le génome de l’AAV2 sauvage est un ADN simple brin composé de trois ORF, dont rep codant les protéines nécessaires à la réplication du virus, et cap codant les protéines de la capside virale. Le troisième ORF est un ORF alternatif dans cap, codant l’AAP, impliquée notamment dans l’encapsidation de l’AAV. Les ITR (inverted terminal repeat) sont des séquences répétées impliquées dans la réplication et l’encapsidation du virus. Dans le génome de l’AAVr, les ORF rep et cap sont remplacées par le gène thérapeutique choisi et un promoteur contrôlant son expression. La production en laboratoire de l’AAV recombinant est possible grâce à l’utilisation d’un plasmide auxiliaire contenant : (1) des gènes adénoviraux, dont les fonctions auxiliaires permettent de maximiser la production du virus, (2) l’ORF rep d’AAV2, et (3) l’ORF cap du sérotype choisi en fonction du tissu ou de l’organe à cibler. L’AAV issu de cette production est alors appelé AAV2/x, x correspondant au sérotype choisi ; néanmoins, par simplification, il est souvent noté AAVx. Cap : capside ; CGM : cellule gliale de Müller ; EPR : épithélium pigmentaire rétinien ; ITR : inverted terminal repeat ; rep : réplication ; SNC : système nerveux central ; PR : photorécepteur. |
Certains vecteurs dérivés des virus adéno-associés sont actuellement les plus prometteurs pour le transfert de gènes thérapeutiques vers la rétine. Récemment, il a été démontré que l’administration sous-rétinienne d’AAV2 est sûre et efficace chez des enfants atteints d’une forme rare de cécité héréditaire, l’amaurose congénitale de Leber, ce qui suggère que la thérapie génique rétinienne par les virus adéno-associés pourrait être étendue avec succès à d’autres affections cécitantes. La grande diversité des virus adéno-associés plaide aussi en ce sens. Il existe en effet un grand nombre de variants capables de cibler différents types de cellules dans la rétine (Tableau III), qu’il s’agisse de cellules gliales, épithéliales, ainsi que de nombreux types de neurones rétiniens.
Les types cellulaires ciblés par différents variants de virus adéno-associés (AAV). En italique sont représentés les AAV obtenus après ingénierie de la capside ; en police normale (romain) sont représentés les AAV avec des capsides naturelles.
Néanmoins, les virus adéno-associés naturels ne permettent pas un ciblage efficace de certains types de cellules. Lorsque les propriétés recherchées ne sont pas présentes dans les sérotypes naturels, l’ingénierie de la capside du virus peut être utilisée pour répondre à nos besoins en thérapie génique.
L’ingénierie de la capside virale des virus adéno-associés
L’ingénierie de la capside d’AAV comporte deux types de méthodes :
-
Le design rationnel
Il consiste à introduire des modifications ciblées pour améliorer les propriétés virales. Nous décrivons ci-dessous deux exemples de mutations pour illustrer ce type d’ingénierie.
-
Mutations ponctuelles de tyrosines, prolines et/ou sérines. La voie ubiquitine-protéasome joue un rôle essentiel dans le trafic intracellulaire des virus adéno-associés. Lors de son transport dans la cellule, la capside virale est sujette à des modifications conformationnelles ; le virus peut être ubiquitiné et dégradé par le système ubiquitine-protéasome. La phosphorylation de certains résidus d’acides aminés exposés en surface de la capside constitue le signal primaire de l’ubiquitination. La suppression de tyrosines (Y) et/ou de sérines (S) critiques permet d’augmenter l’efficacité de transduction par rapport aux vecteurs de type sauvage [29]. Selon de récentes études, la mutagenèse des thréonines (T) exposées en surface permettrait également d’améliorer cette efficacité [30]. Ces observations suggèrent que la poursuite de l’optimisation de la capside du virus adéno-associé, par modification des résidus d’acides aminés impliqués dans la phosphorylation, est une piste importante pour augmenter l’efficacité de transfert de gènes. Une de ces études a ainsi conduit à la génération d’un vecteur (issu de l’AAV2) porteur de quatre mutations de tyrosines [31], ayant un potentiel intéressant pour une utilisation en thérapie génique humaine oculaire.
-
Insertions peptidiques. L’utilisation de vecteurs AAV2 ou issus d’AAV2 est souvent limitée par l’abondance de protéoglycanes à sulfate d’héparane (HSPG), le récepteur primaire du virus, ce qui rend le vecteur « non spécifique ». Afin d’obtenir un transfert de gènes indépendant de ces protéoglycanes, plusieurs groupes ont montré que le tropisme de l’AAV peut être modifié par remaniement génétique de la capside virale en y insérant de courtes séquences peptidiques 32–34.
Le design rationnel vise donc à effectuer des substitutions ou insertions ciblées pour améliorer l’efficacité du transfert génique. Nous pouvons aussi manipuler la capside virale en utilisant des méthodes moléculaires combinatoires, comme l’évolution dirigée, permettant de créer plus de diversité.
-
-
Criblage à haut débit par évolution dirigée
La stratégie de l’évolution dirigée consiste à mimer le processus naturel de l’évolution, mais de façon accélérée et au laboratoire. En vectorologie, elle permet de modifier les propriétés d’un vecteur pour obtenir un phénotype précis (d’où l’expression d’évolution dirigée), par exemple modifier la capside virale pour améliorer le tropisme du virus envers un tissu particulier ou encore obtenir des variants échappant au système immunitaire [33]. Pour cela, le génome du virus est modifié par différents procédés de mutagenèse aléatoire et des millions de variants sont créés, constituant ainsi des librairies hautement diverses. À la suite d’un crible spécialement adapté à l’objectif défini, le ou les meilleurs variants sont sélectionnés parmi les millions qui sont générés. L’évolution dirigée est donc un outil puissant, car elle permet de produire des vecteurs viraux adéno-associés mutants qui ne peuvent, a priori, être prédits par des analyses de la capside virale in silico, ou qui n’auraient peut-être jamais été générés dans le cours de l’évolution naturelle. De plus, le processus de l’évolution dirigée se fait sur un laps de temps court (quelques mois), contrairement à l’évolution naturelle qui requiert des milliers voire des millions d’années.
Dans le cas de la rétine, la stratégie de l’évolution dirigée a été utilisée avec succès et a permis de créer des variants de virus adéno-associés plus performants pour la transduction de la rétine. Un premier exemple de criblage à haut débit d’une librairie de virus adéno-associés se distinguant par leur capside, a permis l’identification de ShH10, un variant viral transduisant spécifiquement les cellules gliales de Müller [35], impliquées, entre autres, dans la neuroprotection de la rétine et constituant un soutien structural. Ce variant a, par la suite, permis de réaliser une thérapie génique avec expression du facteur neurotrophique GDNF (glial cell-line derived neurotrophic factor) dans les cellules gliales de Müller, ce qui a permis de ralentir la dégénérescence de la rétine dans un modèle murin de rétinopathie pigmentaire [36].
Un deuxième exemple de succès de l’utilisation de l’évolution moléculaire pour la création de nouveaux variants de virus adéno-associés a été obtenu en utilisant un processus de sélection in vivo chez le rongeur. Le criblage des librairies de mutants a permis d’isoler AAV2.7m8, un variant pouvant transduire efficacement toutes les couches de la rétine chez le rongeur, et ce par voie intravitréenne, donc sans aucun dommage à la rétine [32]. Les photorécepteurs et l’épithélium rétinien pigmentaire, cellules situées le plus en profondeur dans la rétine, et donc les plus difficiles à atteindre, sont aussi transduites par ce variant. AAV2.7m8 se caractérise par l’insertion d’un peptide de dix acides aminés exposé 60 fois par la capside du virus. L’évolution dirigée a donc ici permis d’obtenir un variant d’AAV plus performant, permettant un ciblage spécifique de cellules rétiniennes (Tableau III) grâce au choix de promoteurs adaptés, pour une expression stable et constante d’un transgène à long terme.
L’utilisation de ces vecteurs doit donc être adaptée à chaque scénario, de nombreux facteurs devant être pris en compte pour optimiser la transduction de la rétine de rongeur, notamment le type de vecteur et le sérotype choisis, mais aussi le promoteur, la voie d’injection, l’état de la rétine (saine ou dégénérée par exemple), etc. À ces facteurs s’ajoute une difficulté supplémentaire : les différences de transduction interespèces.
Défis translationnels
Le tropisme cellulaire des différents vecteurs viraux adéno-associés, ainsi que l’efficacité d’un traitement par thérapie génique, peuvent en effet varier selon le modèle animal. Ainsi, AAV8 est idéal pour transduire les photorécepteurs de la rétine murine, tandis qu’AAV9 est plus efficace qu’AAV8 pour la transduction des photorécepteurs (notamment des cônes) dans la rétine de primates [48, 49]. Afin d’optimiser une thérapie génique chez l’homme, il est donc important de pouvoir tester l’efficacité de différents vecteurs chez des primates non humains.
L’injection sous-rétinienne de vecteurs dérivés de lentivirus ou de virus adéno-associés a été testée chez le primate, et ces deux vecteurs semblent pouvoir cibler l’épithélium pigmentaire sans difficulté. Néanmoins, la transduction étendue de la rétine neurale interne reste problématique chez le primate [32, 50]. L’extension de l’aire d’infection au-delà de la fovéa et des zones vasculaires reste donc un défi pour ces virus adéno-associés. Les différences de transduction interespèces après une injection intra-vitréenne semblent être liées aux différences d’épaisseur de la membrane limitante interne (Figure 2), qui constitue une barrière physique au passage des vecteurs [38].
Conclusion
Pour des transgènes de petite taille, le virus adéno-associé semble donc être le vecteur le plus adapté aux besoins d’un transfert de gènes thérapeutiques dans l’intégralité de la rétine de souris et dans la rétine de primate. Aujourd’hui, le développement des techniques de modification de la capside de ces viurs (à la fois rationnelles et combinatoires), ainsi que la disponibilité d’un grand nombre de promoteurs spécifiques de neurones, de glie ou de l’épithélium pigmentaire rétinien, nous donnent la possibilité de cibler de manière efficace et techniquement reproductible une grande diversité de cellules rétiniennes. Nous avons donc les moyens biotechnologiques pour étudier le fonctionnement de la rétine de mammifère et tester des stratégies de thérapie génique, afin d’apporter des preuves de concept de leur efficacité chez le rongeur. La translation de ces résultats vers l’œil de primate non humain est un des principaux défis de la thérapie génique oculaire à visée clinique, et nous attendons des avancées rapides dans ce domaine dans les années à venir.
Liens d’intérêt
Deniz Dalkara est auteur d’un brevet en lien avec le vecteur AAV2-7m8 mentionné dans cet article (adeno-associated virus with variant capsid and methods of use there of, U.S. application No 61/478,355, WO2012145601). Hanen Khabou déclare n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
« Dystrophie maculaire héréditaire rare d’apparition précoce caractérisée par une perte progressive de la vision centrale, mais avec une vision périphérique intacte, une légère perte de la vision des couleurs, une adaptation à l’obscurité retardée, et une atrophie maculaire avec ou sans taches paramaculaires et une dégénérescence de l’épithélium pigmentaire de la rétine. Liée à la mutation du gène ABCA4 ». (Orphanet).
« Le syndrome d’Usher (SU) se caractérise par une atteinte de l’audition, une perte progressive de la vision secondaire à une rétinite pigmentaire (RP) et dans certains cas par des troubles vestibulaires. Il se transmet comme une affection autosomique récessive et c’est la première cause de surdité associée à une cécité chez l’homme. Le syndrome d’Usher constitue une entité hétérogène au plan clinique et génétique. Au moins 12 loci chromosomiques ont été identifiés jusqu’à présent et assignés aux trois formes cliniques majeures : les formes USH1, USH2 et USH3 ». (reproduit de Institut-vision.org).
Formation d’une particule longue par concaténation de particules plus courtes.
Références
- Wright AF, Chakarova CF, Abd El-Aziz MM, Bhattacharya SS. Photoreceptor degeneration: genetic and mechanistic dissection of a complex trait. Nat Rev Genet 2010 ; 11 : 273–284. [CrossRef] [PubMed] [Google Scholar]
- Bainbridge JWB, Smith AJ, Barker SS, et al. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med 2008 ; 358 : 2231–2239. [CrossRef] [PubMed] [Google Scholar]
- Cideciyan AV, Aleman TS, Boye SL, et al. Human gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision but with slow rod kinetics. Proc Natl Acad Sci USA 2008 ; 105 : 15112–15117. [CrossRef] [Google Scholar]
- Maguire AM, Simonelli F, Pierce EA, et al. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N Engl J Med 2008 ; 358 : 2240–2248. [CrossRef] [PubMed] [Google Scholar]
- Maclaren RE, Groppe M, Barnard AR, et al. Retinal gene therapy in patients with choroideremia: initial findings from a phase 1/2 clinical trial. Lancet 2014 ; 383 : 1129–1137. [CrossRef] [PubMed] [Google Scholar]
- Bainbridge JW, Ali RR. Success in sight: the eyes have it! Ocular gene therapy trials for LCA look promising. Gene Ther 2008 ; 15 : 1191–1192. [CrossRef] [PubMed] [Google Scholar]
- Koirala A, Conley SM, Naash MI. A review of therapeutic prospects of non-viral gene therapy in the retinal pigment epithelium. Biomaterials 2013 ; 34 : 7158–7167. [CrossRef] [PubMed] [Google Scholar]
- Vandenberghe LH, Auricchio A. Novel adeno-associated viral vectors for retinal gene therapy. Gene Ther 2012 ; 19 : 162–168. [CrossRef] [PubMed] [Google Scholar]
- Han Z, Conley SM, Naash MI. AAV and compacted DNA nanoparticles for the treatment of retinal disorders: challenges and future prospects. Invest Ophthalmol Vis Sci 2011 ; 52 : 3051–3059. [CrossRef] [PubMed] [Google Scholar]
- Bennett J, Wilson J, Sun D, et al. Adenovirus vector-mediated in vivo gene transfer into adult murine retina. Invest Ophthalmol Vis Sci 1994 ; 35 : 2535–2542. [PubMed] [Google Scholar]
- Reichel MB, Ali RR, Thrasher AJ, et al. Immune responses limit adenovirally mediated gene expression in the adult mouse eye. Gene Ther 1998 ; 5 : 1038–1046. [CrossRef] [PubMed] [Google Scholar]
- Vollrath D, Feng W, Duncan JL, et al. Correction of the retinal dystrophy phenotype of the RCS rat by viral gene transfer of Mertk. Proc Natl Acad Sci USA 2001 ; 98 : 12584–12589. [CrossRef] [Google Scholar]
- Burcin MM, Schiedner G, Kochanek S, et al. Adenovirus-mediated regulable target gene expression in vivo. Proc Natl Acad Sci USA 1999 ; 96 : 355–360. [CrossRef] [Google Scholar]
- Balaggan KS, Ali RR. Ocular gene delivery using lentiviral vectors. Gene Ther 2012 ; 19 : 145–153. [CrossRef] [PubMed] [Google Scholar]
- Duisit G, Conrath H, Saleun S, et al. Five recombinant simian immunodeficiency virus pseudotypes lead to exclusive transduction of retinal pigmented epithelium in rat. Mol Ther 2002 ; 6 : 446–454. [CrossRef] [PubMed] [Google Scholar]
- Kachi S, Binley K, Yokoi K, et al. Equine infectious anemia viral vector-mediated codelivery of endostatin and angiostatin driven by retinal pigmented epithelium-specific VMD2 promoter inhibits choroidal neovascularization. Hum Gene Ther 2009 ; 20 : 31–39. [CrossRef] [PubMed] [Google Scholar]
- Bemelmans AP, Kostic C, Crippa SV, et al. Lentiviral gene transfer of Rpe65 rescues survival and function of cones in a mouse model of leber congenital amaurosis. PLoS Med 2006 ; 3 : 1892–1903. [CrossRef] [Google Scholar]
- Balaggan KS, Binley K, Esapa M, et al. Stable and efficient intraocular gene transfer using pseudotyped EIAV lentiviral vectors. J Gene Med 2006 ; 8 : 275–285. [CrossRef] [PubMed] [Google Scholar]
- Lipinski D, Barnard A. Venezuelan equine encephalitis virus-derived glycoprotein-pseudotyped lentivirus vectors differentially transduce corneal endothelium, trabecular meshwork. Hum Gene Ther 2013 ; 25 : 50–62. [CrossRef] [PubMed] [Google Scholar]
- Binley K, Widdowson P, Loader J, et al. Transduction of photoreceptors with equine infectious anemia virus lentiviral vectors: safety and biodistribution of StarGen for Stargardt disease. Invest Ophthalmol Vis Sci 2013 ; 54 : 4061–4071. [CrossRef] [PubMed] [Google Scholar]
- Hellström M, Ruitenberg MJ, Pollett MA, et al. Cellular tropism and transduction properties of seven adeno-associated viral vector serotypes in adult retina after intravitreal injection. Gene Ther 2009 ; 16 : 521–532. [CrossRef] [PubMed] [Google Scholar]
- Allocca M, Mussolino C, Garcia-Hoyos M, et al. Novel adeno-associated virus serotypes efficiently transduce murine photoreceptors. J Virol 2007 ; 81 : 11372–11380. [CrossRef] [PubMed] [Google Scholar]
- Nathwani AC, Tuddenham EGD, Rangarajan S, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med 2011 ; 365 : 2357–2365. [CrossRef] [PubMed] [Google Scholar]
- Kay MA, Manno CS, Ragni MV, et al. Evidence for gene transfer and expression of factor IX in haemophilia B patients treated with an AAV vector. Nat Genet 2000 ; 24 : 257–261. [CrossRef] [PubMed] [Google Scholar]
- Büning H, Perabo L, Coutelle O, et al. Recent developments in adeno-associated virus vector technology. J Gene Med 2008 ; 10 : 717–733. [CrossRef] [PubMed] [Google Scholar]
- Nonnenmacher M, Weber T. Intracellular transport of recombinant adeno-associated virus vectors. Gene Ther 2012 ; 19 : 649–658. [CrossRef] [PubMed] [Google Scholar]
- Abraham D, Handler C, Dashwood M, Coghlan G. Advances in vascular medicine. Google eBook : Springer, 2010. http://books.google.com/books?id=EYp_Wa6CKtkC&pgis=1 [CrossRef] [Google Scholar]
- Nonnenmacher M, Weber T. Intracellular transport of recombinant adeno-associated virus vectors. Gene Ther 2012 ; 19 : 649–658. [CrossRef] [PubMed] [Google Scholar]
- Zhong L, Li B, Mah CS, et al. Next generation of adeno-associated virus 2 vectors: point mutations in tyrosines lead to high-efficiency transduction at lower doses. Proc Natl Acad Sci USA 2008 ; 105 : 7827–7832. [CrossRef] [Google Scholar]
- Kay CN, Ryals RC, Aslanidi GV, et al. Targeting photoreceptors via intravitreal delivery using novel, capsid-mutated AAV vectors. PLoS One 2013 ; 8 : e62097. [CrossRef] [PubMed] [Google Scholar]
- Petrs-Silva H, Dinculescu A, Li Q, et al. Novel properties of tyrosine-mutant AAV2 vectors in the mouse retina. Mol Ther 2011 ; 19 : 293–301. [CrossRef] [PubMed] [Google Scholar]
- Dalkara D, Byrne LLC, Klimczak RR, et al. In vivo-directed evolution of a new adeno-associated virus for therapeutic outer retinal gene delivery from the vitreous. Sci Transl Med 2013 ; 5 : 189ra176. [CrossRef] [Google Scholar]
- Maheshri N, Koerber JT, Kaspar BK, Schaffer DV. Directed evolution of adeno-associated virus yields enhanced gene delivery vectors. Nat Biotechnol 2006 ; 24 : 198–204. [CrossRef] [PubMed] [Google Scholar]
- Müller OJ, Kaul F, Weitzman MD, et al. Random peptide libraries displayed on adeno-associated virus to select for targeted gene therapy vectors. Nat Biotechnol 2003 ; 21 : 1040–1046. [CrossRef] [PubMed] [Google Scholar]
- Klimczak RR, Koerber JT, Dalkara D, et al. A novel adeno-associated viral variant for efficient and selective intravitreal transduction of rat Müller cells. PLoS One 2009 ; 4 : e7467. [CrossRef] [PubMed] [Google Scholar]
- Dalkara D, Kolstad KD, Guerin KI, et al. AAV mediated GDNF secretion from retinal glia slows down retinal degeneration in a rat model of retinitis pigmentosa. Mol Ther 2011 ; 19 : 1602–1608. [CrossRef] [PubMed] [Google Scholar]
- Yin L, Greenberg K, Hunter JJ, et al. Intravitreal injection of AAV2 transduces macaque inner retina. Invest Ophthalmol Vis Sci 2011 ; 52 : 2775–2783. [CrossRef] [PubMed] [Google Scholar]
- Dalkara D, Kolstad KD, Caporale N, et al. Inner limiting membrane barriers to AAV-mediated retinal transduction from the vitreous. Mol Ther 2009 ; 17 : 2096–2102. [CrossRef] [PubMed] [Google Scholar]
- Petrs-Silva H, Dinculescu A, Li Q, et al. High-efficiency transduction of the mouse retina by tyrosine-mutant AAV serotype vectors. Mol Ther 2009 ; 17 : 463–471. [CrossRef] [PubMed] [Google Scholar]
- Doroudchi MM, Greenberg KP, Liu J, et al. Virally delivered channelrhodopsin-2 safely and effectively restores visual function in multiple mouse models of blindness. Mol Ther 2011 ; 19 : 1220–1229. [CrossRef] [PubMed] [Google Scholar]
- Dorrell MI, Aguilar E, Jacobson R, et al. Antioxidant or neurotrophic factor treatment preserves function in a mouse model of neovascularization-associated oxidative stress. J Clin Invest 2009 ; 119 : 611–623. [CrossRef] [PubMed] [Google Scholar]
- Koerber JT, Klimczak R, Jang JH, et al. Molecular evolution of adeno-associated virus for enhanced glial gene delivery. Mol Ther 2009 ; 17 : 2088–2095. [CrossRef] [PubMed] [Google Scholar]
- Auricchio A, Kobinger G, Anand V, et al. Exchange of surface proteins impacts on viral vector cellular specificity and transduction characteristics: the retina as a model. Hum Mol Genet 2001 ; 10 : 3075–3081. [CrossRef] [PubMed] [Google Scholar]
- Petrs-Silva H, Dinculescu A, Li Q, et al. Novel properties of tyrosine-mutant AAV2 vectors in the mouse retina. Mol Ther 2009 ; 19 : 293–301. [Google Scholar]
- Natkunarajah M, Trittibach P, McIntosh J, et al. Assessment of ocular transduction using single-stranded and self-complementary recombinant adeno-associated virus serotype 2/8. Gene Ther 2008 ; 15 : 463–467. [CrossRef] [PubMed] [Google Scholar]
- Weber M, Rabinowitz J, Provost N, et al. Recombinant adeno-associated virus serotype 4 mediates unique and exclusive long-term transduction of retinal pigmented epithelium in rat, dog, and nonhuman primate after subretinal delivery. Mol Ther 2003 ; 7 : 774–781. [CrossRef] [PubMed] [Google Scholar]
- Allocca M, Doria M, Petrillo M, et al. Serotype-dependent packaging of large genes in adeno-associated viral vectors results in effective gene delivery in mice. J Clin Invest 2008 ; 118 : 1955–1964. [CrossRef] [PubMed] [Google Scholar]
- Bennett J, Maguire AM, Cideciyan AV, et al. Stable transgene expression in rod photoreceptors after recombinant adeno-associated virus-mediated gene transfer to monkey retina. Proc Natl Acad Sci USA 1999 ; 96 : 9920–9925. [CrossRef] [Google Scholar]
- Vandenberghe LH, Bell P, Maguire AM, et al. AAV9 targets cone photoreceptors in the nonhuman primate retina. PLoS One 2013 ; 8 : e53463. [CrossRef] [PubMed] [Google Scholar]
- Yin L, Greenberg K, Hunter JJ, et al. Intravitreal injection of AAV2 transduces macaque inner retina. Invest Ophthalmol Vis Sci 2011 ; 52 : 2775–2783. [CrossRef] [PubMed] [Google Scholar]
Liste des tableaux
Caractéristiques des trois principaux vecteurs viraux utilisés en thérapie génique oculaire. kb : kilobases.
Récepteurs cellulaires de différents sérotypes d’AAV 25–27. EGFR : epidermal growth factor receptor ; FGF : fibroblast growth factor ; PDGF : platelet-derived growth factor.
Les types cellulaires ciblés par différents variants de virus adéno-associés (AAV). En italique sont représentés les AAV obtenus après ingénierie de la capside ; en police normale (romain) sont représentés les AAV avec des capsides naturelles.
Liste des figures
|
Figure 1. L’anatomie de l’œil humain et les voies d’injection intra-oculaires. L’œil humain focalise la lumière sur la rétine (en rose) à l’aide du cristallin, notamment sur la fovéa, la structure responsable de la vision à haute acuité. L’information lumineuse reçue est convertie en signaux électriques, puis transmise au cerveau par le nerf optique. L’injection sous-rétinienne consiste à déposer le vecteur dans l’espace sous-rétinien, entre la rétine et l’épithélium sous-jacent (EPR), directement en contact avec la rétine. Cette voie d’administration entraîne un décollement transitoire de la rétine qui se résorbe par la suite. L’injection intra-vitréenne amène, quant à elle, le vecteur dans le corps vitré, où il va ensuite diffuser et atteindre la rétine. |
| Dans le texte | |
|
Figure 2. Structure de l’œil et de la rétine de souris. L’œil de souris est constitué d’un large cristallin et d’un petit volume de vitré (plus aqueux que le vitré de l’œil humain). La rétine est composée de plusieurs types neuronaux : les neurones de premier ordre sont les photorécepteurs (les cônes et les bâtonnets) ; les neurones de second ordre sont les cellules bipolaires, amacrines et horizontales ; et les cellules ganglionnaires sont les neurones de troisième ordre. L’épithélium pigmentaire (EPR) se situe derrière la rétine neurale, en contact avec les photorécepteurs. Les cellules gliales de Müller traversent verticalement la rétine, leurs pieds étant, d’une part, apposés au corps vitré et secrétant un réseau dense de polysaccharides (formant ainsi la membrane limitante interne [MLI]) et, d’autre part, apposés aux segments internes des photorécepteurs. |
| Dans le texte | |
|
Figure 3. Production de vecteurs AAV recombinants (AAVr) à partir d’AAV2. Le génome de l’AAV2 sauvage est un ADN simple brin composé de trois ORF, dont rep codant les protéines nécessaires à la réplication du virus, et cap codant les protéines de la capside virale. Le troisième ORF est un ORF alternatif dans cap, codant l’AAP, impliquée notamment dans l’encapsidation de l’AAV. Les ITR (inverted terminal repeat) sont des séquences répétées impliquées dans la réplication et l’encapsidation du virus. Dans le génome de l’AAVr, les ORF rep et cap sont remplacées par le gène thérapeutique choisi et un promoteur contrôlant son expression. La production en laboratoire de l’AAV recombinant est possible grâce à l’utilisation d’un plasmide auxiliaire contenant : (1) des gènes adénoviraux, dont les fonctions auxiliaires permettent de maximiser la production du virus, (2) l’ORF rep d’AAV2, et (3) l’ORF cap du sérotype choisi en fonction du tissu ou de l’organe à cibler. L’AAV issu de cette production est alors appelé AAV2/x, x correspondant au sérotype choisi ; néanmoins, par simplification, il est souvent noté AAVx. Cap : capside ; CGM : cellule gliale de Müller ; EPR : épithélium pigmentaire rétinien ; ITR : inverted terminal repeat ; rep : réplication ; SNC : système nerveux central ; PR : photorécepteur. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.