")
")
| Issue |
Med Sci (Paris)
Volume 29, Number 11, Novembre 2013
Le réseau international des Instituts Pasteur
|
|
|---|---|---|
| Page(s) | 1034 - 1041 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/20132911021 | |
| Published online | 20 novembre 2013 | |
Le réseau international des Instituts Pasteur (3)
Éradication de la poliomyélite et émergence de poliovirus pathogènes dérivés du vaccin
De Madagascar au Cameroun
Eradication of poliomyelitis and emergence of pathogenic vaccine-derived polioviruses: from Madagascar to Cameroon
1
Institut Pasteur, biologie des virus entériques, Inserm U994, 25, rue du Docteur Roux, 75724 Paris Cedex 15, France
2
Institut Pasteur de Madagascar, Antananarivo, Madagascar
3
Centre Pasteur du Cameroun, Yaoundé, Cameroun
4
Institut Pasteur de La Guyane, Cayenne, Guyane Française
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
**
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Les campagnes de vaccination visant à éradiquer la poliomyélite sont effectuées avec le vaccin polio oral, un vaccin vivant constitué de souches de poliovirus atténuées des trois sérotypes. Ces campagnes ont permis de faire régresser, puis disparaître la maladie due aux souches sauvages de virus dans la plupart des pays. Cependant, lorsque la couverture vaccinale est faible, les souches vaccinales de poliovirus peuvent circuler dans les populations insuffisamment vaccinées et redeviennent ainsi pathogènes par mutation et par recombinaison génétique avec d’autres souches d’entérovirus. Les souches mutées et recombinantes sont à l’origine d’épidémies de poliomyélite paralytique aiguë. Le phénomène de plasticité génétique qui menace les bénéfices de la vaccination fait l’objet d’une surveillance particulière et est étudié dans les laboratoires de virologie du réseau international des Instituts Pasteur.
Abstract
The oral poliovaccine, a live vaccine made of attenuated poliovirus strains, is the main tool of the vaccination campaigns organised for eradicating poliomyelitis. these campaigns had led to the decline and, thereafter, to the disappearance of wild poliovirus strains of the three serotypes (1-3) in most parts of the world. However, when the poliovaccine coverage becomes too low, vaccine polioviruses can circulate in insufficiently immunized populations and become then pathogenic by mutations and genetic recombination with other enteroviruses of the same species, in particular some coxsackievirus A. These mutated and recombinant vaccine strains have been implicated in several epidemics of paralytic poliomyelitis. Two polio outbreaks associated with these pathogenic circulating vaccine-derived poliovirus (cVDPV) occurred in 2001-2002 and 2005 in the South of Madagascar where vaccine coverage was low. These cVDPV, of serotype 2 or 3, were isolated from paralyzed children and some of their healthy contacts. Other cVDPV were isolated in the same region from healthy children in 2011, indicating that these viruses were circulating again. Vaccination campaigns could stop the outbreaks in 2002 and 2005, and most probably prevent another one in 2011. Therefore, the genetic plasticity of poliovaccine strains that threatens the benefit of vaccination campaigns is the target of an accurate surveillance and an important theme of studies in the virology laboratories of the Institut Pasteur international network.
© 2013 médecine/sciences – Inserm

Le poliovirus, la poliomyélite paralytique et le programme d’éradication
Après le succès mondial de l’éradication de la variole, l’Organisation mondiale de la santé (OMS) a lancé en 1988 le programme d’éradication de la poliomyélite paralytique aiguë (PPA), une maladie essentiellement causée par le poliovirus (PV) [1]. Le programme est basé sur des campagnes massives de vaccination antipoliomyélitique et sur la surveillance des paralysies flasques aiguës (PFA)1. L’Institut Pasteur est impliqué dans les recherches sur le PV depuis le début du XXe siècle. Aujourd’hui, il poursuit ces recherches et participe également à la surveillance du PV dans le cadre d’un réseau qui regroupe les laboratoires de virologie de 12 Instituts Pasteur (réseau entérovirus des Instituts Pasteur, REIP). Ces laboratoires font, en majorité, partie du réseau mondial de l’OMS qui rassemble près de 150 laboratoires chargés de la surveillance de la poliomyélite. Malgré les succès du programme d’éradication (seuls trois pays restent endémiques pour la maladie), les étapes ultimes s’avèrent complexes, avec des stratégies de vaccination ainsi que des méthodes de surveillance en permanente évolution grâce aux données de la recherche [2].
Le PV, dont il existe trois sérotypes (1 à 3), appartient au genre Enterovirus [27] et à la famille des Picornaviridae [3]. Les entérovirus humains comprennent quatre espèces (HEV-A à -D) et les rhinovirus, agents du rhume. Le PV appartient à l’espèce HEV-C, comme plusieurs coxsackie virus A (CVA). Le génome du PV est un ARN à simple brin de polarité positive d’environ 7,5 kilobases, entouré d’une capside de symétrie icosaédrique, composée des quatre protéines virales (VP1 à VP4). Le génome viral comporte une longue phase de lecture ouverte unique, dont la moitié 5’ code pour les protéines de la capside, tandis que la moitié 3’ code pour les protéines non structurales impliquées dans la réplication et la maturation des protéines virales. La longue phase de lecture ouverte est entourée de deux régions 5’ et 3’ non codantes comprenant des signaux de régulation de la réplication, aux deux extrémités, et de la traduction, en 5’ [4].
Le seul hôte naturel du PV est l’homme, et la transmission du virus se fait essentiellement par voie féco-orale. La multiplication du virus commence dans le tube digestif, au niveau de l’oropharynx et de l’intestin grêle. À partir de ces sites, le virus atteint d’abord les ganglions lymphatiques cervicaux et mésentériques, puis la circulation sanguine où s’établit une virémie. Lorsque celle-ci est de grande amplitude, le virus peut atteindre le système nerveux central (SNC) dans lequel il cause directement la mort des motoneurones dans les cornes antérieures de la moelle épinière, le tronc cérébral et le cortex moteur, principalement. Le plus souvent, moins de 1 % des personnes infectées par le PV présentent une PPA. Parmi les HEV-C, seul le PV est un pathogène sévère, et il utilise un récepteur différent des autres virus de la même espèce (CD155 pour le PV au lieu de ICAM-1 [intercellular adhesion molecule-1] pour de nombreux HEV-C). À quelques exceptions près, les CVA de l’espèce C sont non ou peu pathogènes [3].
Les vaccins contre la poliomyélite paralytique
Pour prévenir la PPA, on utilise aujourd’hui deux vaccins très efficaces développés dans les années 1950 (Encadré 1). Le premier, le vaccin polio inactivé injectable (VPI), a été mis au point par J. Salk ; il est composé de souches sauvages inactivées des trois sérotypes. Le second, le vaccin polio oral (VPO), a été mis au point par A. Sabin ; il est composé de souches atténuées des trois sérotypes [4]. Chacun des deux vaccins présente des avantages et quelques inconvénients, mais ils sont tous les deux d’une grande efficacité. Le VPI, utilisé dans la majorité des pays développés, confère une bonne immunité générale et est parfaitement sûr. Cependant, il doit être administré par du personnel médical, nécessite des rappels réguliers, et il est relativement onéreux. Le VPO peut être administré par du personnel non médical, ce qui facilite grandement son utilisation dans les pays en développement. Il est peu onéreux et confère une bonne immunité générale et intestinale en raison de la multiplication des souches dans l’intestin grêle. De ce fait, il est généralement admis qu’il bloque la propagation des PV sauvages. Cette dernière qualité en a fait l’outil choisi par l’OMS pour atteindre l’objectif d’éradiquer la PPA due aux PV sauvages. Le nombre de cas de PPA, estimé à plus de 600 000 dans la période prévaccination, puis à environ 350 000 en 1988, a diminué progressivement jusqu’en 2000 (réduction de 99 %) [2]. Depuis, le nombre de cas se maintient à quelques centaines par an. La maladie a été éradiquée récemment en Inde, et elle n’est plus endémique que dans trois pays du monde : le Nigéria, le Pakistan et l’Afghanistan. La disparition complète des souches sauvages de type 2 dans le monde et l’éradication de la PPA due aux virus sauvages sur plusieurs continents ont montré que l’objectif fixé par l’OMS peut être atteint. Malheureusement, les souches vaccinales Sabin du VPO présentent une certaine instabilité génétique qui est à l’origine de la réversion des déterminants d’atténuation lors de la multiplication de ces souches dans l’intestin. Ces réversions sont à l’origine des très rares cas de PPA qui affectent les enfants en cours de vaccination, ou leurs contacts non vaccinés [5].
1.
La réponse humorale de l’hôte joue un rôle prépondérant dans l’immunité contre le poliovirus. Il existe trois sérotypes - ou types - de poliovirus (1, 2 et 3), et c’est la capside virale, composée des quatre protéines de structures VP1-VP4, qui définit le sérotype. Lorsqu’un individu est infecté par un PV d’un sérotype donné, ou vacciné contre celui-ci, il est ultérieurement protégé contre les poliovirus du même sérotype, grâce à la synthèse d’anticorps neutralisants, mais il ne l’est pas contre les virus des deux autres sérotypes. Il faut donc être vacciné contre les PV des trois sérotypes pour être aujourd’hui complètement protégé contre la poliomyélite paralytique.
L’émergence des souches circulantes de poliovirus dérivées du vaccin (cPVDV)
Dans les pays où la couverture vaccinale n’est pas optimale, une autre conséquence de l’instabilité génétique des souches du VPO est l’émergence de souches circulantes de PV dérivées du vaccin (cPVDV) [6]. La durée de circulation des souches est évaluée d’après une horloge moléculaire basée sur le nombre de mutations qui s’accumulent dans la région génomique codant pour la protéine de capside VP1. Il est à l’heure actuelle admis que les souches vaccinales ayant accumulé au moins 1 % de mutations dans la région codant pour VP1 pour les types 1 et 3 (neuf mutations), et 0,6 % pour le type 2 (six mutations) sont des cPVDV, car ces caractéristiques génétiques indiquent une période de circulation interhumaine (et/ou de multiplication) anormalement longue. Une vingtaine d’épidémies de PPA dues à des cPVDV ont été décrites depuis l’an 20002. Les deux premières ont été causées par des virus de type 1 sur l’île d’Hispaniola (à Saint Domingue et Haïti) et aux Philippines [7, 8]. Une autre épidémie, causée par des virus de type 2, survenue entre 1983 et 1993 en Égypte, a été découverte rétrospectivement [9]. Outre des taux élevés de mutations dans les séquences génomiques vaccinales, les cPVDV ont très généralement la particularité d’être recombinants : leur génome est, pour la région codant pour les protéines de capside, d’origine vaccinale, tandis que la région codant pour les protéines non structurales, et parfois une ou les deux régions non codantes, proviennent d’autres entérovirus de la même espèce HEV-C [6]. L’émergence de ces virus recombinants implique que le même hôte ait été infecté par le PV et un autre HEV-C, et que les deux virus se soient multipliés simultanément dans les mêmes cellules-hôtes du tractus digestif. La caractérisation de ces cPVDV indique qu’ils ont récupéré deux des propriétés biologiques les plus importantes des virus sauvages : la capacité d’être transmis d’homme à homme de manière efficace et la capacité de provoquer des paralysies sévères. Ces données ont obligé l’OMS à reconsidérer ses stratégies d’immunisation contre la poliomyélite et sa méthode de surveillance du PV, basée à l’origine sur la recherche exclusive des PV sauvages dans les selles de tous les enfants de moins de 15 ans atteints de paralysie flasque aiguë (PFA).
Il est généralement admis qu’un taux de couverture vaccinale supérieure à 80 % est requis pour empêcher la propagation des souches sauvages de PV, et il est probable qu’un taux similaire est requis pour empêcher celle des cPVDV. De nombreux pays en développement, qui ont éradiqué la poliomyélite due aux souches de PV sauvages locales grâce à la vaccination, dont certains pays du réseau entérovirus des Instituts Pasteur (REIP), rencontrent des difficultés à maintenir une couverture vaccinale de haut niveau contre cette maladie. Leurs ressources limitées les amènent à négliger une maladie qui a disparu de leur pays depuis plusieurs années, alors qu’ils ont à faire face quotidiennement à d’autres maladies infectieuses redoutables. Or, une mauvaise couverture vaccinale contre la poliomyélite permet, d’une part, l’importation de PV sauvages à partir des rares pays encore endémiques et, d’autre part, la circulation interhumaine, ainsi que la dérive, des virus vaccinaux. Aussi, une attention particulière a-t-elle été portée à la surveillance dans le REIP, avec la mise en place dès 1988 de programmes complémentaires comprenant des tests moléculaires visant à surveiller la dérive des souches vaccinales [10]. C’est grâce à ces tests que la résurgence de cas de poliomyélite à Madagascar en 2001 a pu être associée à la présence de cPVDV [11, 12].
Les épidémies dues aux cPVDV à Madagascar
Dans l’île de Madagascar, la poliomyélite due aux souches sauvages de PV a disparu depuis 1998, grâce à l’organisation de journées nationales de vaccination avec le vaccin polio oral (VPO) en 1997, 1998 et 1999. Cependant, du fait des difficultés à mettre en place et maintenir dans le temps une couverture vaccinale de routine optimale sur certaines régions de l’île, et donc à maintenir une immunité suffisante dans la population, cinq cas de paralysie flasque aiguë associés à des cPVDV ont été découverts quatre ans plus tard, entre octobre 2001 et avril 2002 (Figure 1) [12]. Ces cas sont survenus dans une province au sud de Madagascar chez des enfants âgés de 6 mois à 11 ans. Le premier cas est apparu dans le district urbain de Toliara, les autres dans la commune rurale du district de Taolagnaro, à 400 km à l’est de Toliara. Aucun de ces enfants n’avait reçu toutes les doses d’une vaccination complète, et les cas sont apparus dans des communautés où le taux de couverture vaccinale par le VPO trivalent variait de 7 à 40 % seulement. Des cPVDV, tous de type 2, ont été isolés chez les cinq enfants. L’analyse de la région codant pour les protéines de capside dans la moitié 5’ du génome a montré que ces virus dérivaient de la souche Sabin 2 du VPO, et se partageaient en deux groupes selon leur lieu d’isolement [12, 13]. Dans la région codant pour la protéine de capside VP1, la séquence du génome des cPVDV diverge de celle de la souche parentale Sabin 2 au niveau de 1 % et 2,5 % des nucléotides selon le groupe, ce qui suggère que les souches se sont multipliées chez un ou plusieurs individus pendant 1 et 2,5 ans, respectivement. Les souches de Taolagnaro sont génétiquement très proches les unes des autres, mais diffèrent de la souche de Toliara par 2,9 % des nucléotides, indiquant l’existence de deux lignages génétiques. Par ailleurs, le séquençage des génomes des cPVDV, dans leur région 3’, a permis de mettre en évidence des séquences apparentées à celles des derniers PV sauvages ayant circulé sur l’île et à celles de CVA (coxsackie virus A) de la même espèce (HEV-C) (Figure 2). Les derniers PV sauvages ayant disparu de l’île depuis plusieurs années, l’hypothèse la plus probable était donc que les cPVDV malgaches étaient issus d’événements de recombinaison entre la souche Sabin 2 du VPO, mutée, et un ou plusieurs CVA [13]. Il est d’ailleurs intéressant de remarquer que le PV et ces CVA ont été reclassés dans la même espèce tout récemment, en raison de leur parenté génétique et de la mise en évidence d’événements de recombinaison. Deux autres cPVDV, semblables à ceux isolés des cas de paralysie flasque aiguë, ont été isolés chez des enfants sains lors d’une collecte de selles effectuée dans la même région de l’île, peu après la riposte vaccinale destinée à juguler l’épidémie.
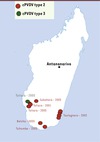 |
Figure 1. Localisation des cas de poliomyélite dus à des cPVDV de type 2 ou 3 à Madagascar en 2001, 2002 et 2005. Chaque point représente un cas de poliomyélite et probablement 100 à 1 000 personnes infectées mais dépourvues de symptômes. Le nom du district et l’année où le cas a été rapporté sont indiqués. La localisation de la capitale du Pays, Antananarivo, est également mentionnée. |
 |
Figure 2. Structure schématique des génomes de différents cPVDV recombinants isolés à Madagascar de 2001 à 2011. L’organisation génomique du poliovirus indique les différentes régions 5’ et 3’ non codantes (5’ NC et 3’ NC), ainsi que la phase ouverte de lecture codant pour les protéines structurales (en bleu) et non structurales (en jaune et en vert). La longueur du génome est indiquée (une unité = 103 nucléotides). L’origine des séquences constituant les génomes recombinants est indiquée selon qu’elle se réfère à des séquences d’origine vaccinale (Sabin) mutées ou à des séquences d’autres HEV-C. Les séquences se rapprochant de celles de souches de virus coxsackie connues sont indiquées. |
La présence quasi simultanée des deux lignages de cPVDV à Madagascar en 2001-2002 témoigne de la fréquence des recombinaisons entre les souches du VPO et d’autres HEV-C. De fait, en dépit de campagnes nationales de vaccination organisées en septembre et octobre 2002 pour interrompre la transmission des cPVDV, une autre épidémie s’est produite dans la même province moins de trois ans plus tard, d’avril à août 2005 [14]. Des cPVDV de type 2 et de type 3 (dérivés respectivement des souches vaccinales Sabin 2 et Sabin 3) ont causé cette nouvelle épidémie. Les génomes des cPVDV de type 2 de 2005 présentent des extrémités 5’ semblables, avec des séquences HEV-C et des sites de recombinaison similaires [15]. De plus, certains génomes de cPVDV de type 2 et de type 3 ont des extrémités 3’ recombinantes analogues présentant des séquences similaires et des sites de recombinaison identiques. Des génomes recombinants quadripartites ont également été observés (Figure 2). Par ailleurs, nous avons trouvé que 7 % des enfants sains vivant à proximité des enfants paralysés hébergeaient des cPVDV qui avaient circulé de 12 à 32 mois d’après l’horloge moléculaire de leur génome. Tous ces virus ont un génome recombinant entre PV et HEV-C, confirmant qu’émergence des cPVDV et co-circulation des HEV-C vont de pair à Madagascar [14, 15].
Alors que les HEV-C sont peu fréquents en France comme dans une grande partie de l’Europe, ils ont été fréquemment détectés parmi les entérovirus isolés à partir des selles d’enfants sains à Madagascar, dans la région où des cPVDV ont émergé. Ainsi, 21 % des selles d’enfants sains testées se sont avérées positives pour les HEV, parmi lesquels 80 % appartiennent à l’espèce HEV-C. Cela suggère que certains des enfants sont co-infectés par deux virus HEV-C différents. Six types différents ont été trouvés : CVA-11, -13, -17, -20, -24 et HEV-99 [16]. Le séquençage du génome de ces virus dans les régions des protéines non structurales suggère que les cPVDV de 2002 ont des régions génomiques apparentées à celles de souches locales de CVA-17 et CVA-13. Les cPVDV de 2005 ont également des régions non structurales apparentées à ces deux types de CVA ; des séquences de CVA-11 pourraient être impliquées dans la recombinaison de manière ponctuelle [15]. Les mécanismes moléculaires qui aboutissent à la formation des cPVDV recombinants restent à élucider. L’analyse du génome de ces virus suggère qu’il s’agit de recombinaison homologue. Cependant, l’hypothèse d’une recombinaison non homologue précédant la recombinaison homologue ne peut être exclue.
Depuis l’épidémie de poliomyélite de 2005, aucun nouveau cas de paralysie associé à des cPVDV n’a été mis en évidence à Madagascar. Cependant, en juin 2011, une nouvelle étude, ayant pour objectif d’étudier la circulation des entérovirus dans la province de Toliara, a permis de mettre en évidence de nouveaux cPVDV circulant chez des enfants sains [17]. Cette étude a consisté à rechercher les entérovirus présents dans les selles d’enfants bien-portants (non paralysés) de moins de cinq ans n’ayant pas reçu de VPO pendant les 30 jours précédant la collecte. Parmi les 616 échantillons de selles collectés, trois cPVDV de type 2 provenant d’enfants différents ont été isolés. Les analyses génomiques de ces virus ont montré qu’ils avaient accumulé, pour l’un, 0,6 % de mutations, et pour les deux autres, autour de 3 % dans la région codant pour la protéine VP1. Ce taux de mutations indiquait qu’ils avaient circulé dans la population pendant six mois et trois ans, respectivement. Par ailleurs, ces analyses ont montré que le virus le plus récent possédait un génome en tous points dérivé du vaccin, alors que les deux autres étaient des virus recombinants entre PV vaccinal et d’autres HEV-C [17]. La découverte de ces virus indiquait une circulation anormale des virus vaccinaux et donc une couverture vaccinale contre la poliomyélite trop faible, ce qui a permis d’alerter les autorités de santé publique. La collaboration entre les autorités malgaches et les responsables régionaux de l’OMS et de l’UNICEF (United nations international children’s emergency fund) a abouti à la mise en place rapide de campagnes de vaccination « porte-à-porte » dans les régions où les cPVDV circulaient. Jusqu’à ce jour, aucun cas de paralysie n’a été rapporté suggérant que les campagnes de vaccination ont permis de limiter, sinon de stopper la circulation de ces souches d’origine vaccinale potentiellement pathogènes.
La découverte de nouvelles souches de cPVDV recombinantes dans la province sud de Madagascar laisse penser qu’une faible couverture vaccinale n’est pas le seul facteur de risque d’épidémie à cPVDV, et que la présence d’autres HEV-C co-circulants pourrait favoriser l’émergence des cPVDV. L’ensemble des résultats obtenus suite à l’étude des cPVDV malgaches montrent une co-évolution rapide des PV et HEV-C, avec de multiples recombinaisons intra- et intertypiques (Encadré 2), sur une courte période et dans une zone géographique restreinte. Les CVA-17 et CVA-13 seraient des partenaires privilégiés de recombinaison avec le PV. Nos études in vitro ont confirmé que ces virus sont des partenaires de recombinaison compatibles [18]. Il n’est pas possible, à l’heure actuelle, d’exclure totalement l’hypothèse selon laquelle la recombinaison intertypique chez les HEV-C serait une conséquence neutre de la présence simultanée du PV et de plusieurs autres HEV-C, mais cette hypothèse paraît de moins en moins probable [19]. Il faut aussi remarquer que les déterminants majeurs de l’atténuation des souches vaccinales Sabin, qui se trouvent dans la région 5’ non codante et affectent l’efficacité de la traduction, ont subi, chez les cPVDV, des réversions vers le génotype sauvage [13, 15, 20]. Par ailleurs, de nombreux cPVDV ont acquis une région 5’ non codante provenant d’HEV-C non-PV, ce qui suggère que la recombinaison peut contribuer de manière indirecte à la réversion des déterminants d’atténuation situés dans cette région. De même, certains codons de la capside, connus pour jouer un rôle dans l’atténuation des souches Sabin, sont mutés chez ces virus. Ces modifications génétiques peuvent expliquer, au moins en partie, le caractère neurovirulent des cPVDV que l’on peut constater lorsqu’ils sont inoculés à des souris transgéniques pour le récepteur humain du PV (CD155). Ces souris développent en effet une maladie similaire à celle de l’homme lorsqu’on leur inocule des PV pathogènes par voie parentérale ou intranasale. Il est intéressant de noter que des cPVDV recombinants très pathogènes émergent à partir de virus non ou peu pathogènes : les souches vaccinales de PV et les CVA de l’espèce C. Par ailleurs, l’antigénicité de ces cPVDV est parfois légèrement modifiée par rapport à celle des souches vaccinales mais, jusqu’à présent, tous les cPVDV décrits peuvent être neutralisés par les anticorps protecteurs présents chez les individus vaccinés, et les épidémies de cPVDV sont arrêtées par des campagnes de vaccination avec le VPO [6, 15].
2.
Un virus recombinant intratypique est le produit de la recombinaison entre deux ou plusieurs virus de même sérotype (par exemple : PV souche sauvage de type 1/PV souche Sabin 1), alors qu’un virus recombinant intertypique est le produit de la recombinaison entre deux ou plusieurs virus de sérotypes différents (par exemple : PV souche Sabin 2/ PV souche Sabin 3, ou PV souche Sabin 2/CVA 17). Il est admis que lors de recombinaisons entre HEV, ceux-ci appartiennent à la même espèce (ici HEV-C).
Les autres épidémies dans le monde
Une vingtaine d’épidémies à cPVDV sont apparues ces dernières années dans diverses régions du monde, touchant un nombre restreint d’individus. Cependant, une épidémie à cPVDV de type 2 a sévi au Nigéria de 2005 à 2012 avec presque 400 cas de PPA [21]. Des divergences nucléotidiques allant jusqu’à 6 % au niveau de la région codant VP1 ont été observées dans le génome des souches nigérianes, confirmant une très longue circulation, et comme dans l’immense majorité des épidémies, les cPVDV impliqués ont un génome recombinant. Une lignée de cPVDV non recombinants, mise en évidence dans deux cas de paralysie flasque aiguë, a néanmoins été détectée en Chine. Beaucoup de ces épidémies se sont produites dans des régions tropicales, comme à Madagascar ou au Nigéria, mais certaines ont eu lieu en Afghanistan, au Pakistan, ou au Yemen, indiquant que le climat tropical humide n’est pas un paramètre déterminant dans l’émergence des cPVDV. En revanche, un écosystème caractérisé par une fréquence et une diversité élevées des HEV-C, notamment les CVA-13 et CVA-17, créerait des conditions particulièrement favorables à l’émergence et à la circulation des souches de cPVDV recombinantes pathogènes [20].
C’est cette hypothèse qui a justifié un travail collaboratif concernant tous les pays du REIP, ayant pour objectif de savoir si des écosystèmes viraux similaires à celui du sud de Madagascar seraient retrouvés dans d’autres régions du monde et, dans l’affirmative, s’ils seraient associés à la circulation de cPVDV. Les résultats obtenus jusqu’à maintenant, concernant quatre pays, indiquent que les HEV-C circulent très peu en Roumanie et en Tunisie [22, 23]. En revanche, des fréquences relativement élevées ont été mises en évidence en République Centrafricaine et au Cameroun [24, 25].
Au Cameroun, nous nous sommes intéressés plus particulièrement à la circulation et à la diversité génétique des HEV circulant chez les enfants sains dans l’extrême nord du pays, une région limitrophe des états du Nigéria où l’émergence de cPVDV est récurrente [21]. Presque 37 % des enfants se sont révélés porteurs d’HEV non PV et, parmi ceux qui étaient positifs, plus de 63 % étaient infectés par des HEV-C [25]. Cette fréquence élevée des HEV-C était associée à une grande diversité génétique, indiquant une endémicité importante de cette espèce d’entérovirus dans la région. En particulier, le CVA-13 avait la plus grande prévalence, et une extrême diversité intratypique. Cet écosystème viral, similaire à celui du sud de Madagascar, est présent au Tchad et, très probablement aussi, dans le nord du Nigeria. En effet, les mouvements des populations sont permanents à travers les frontières du Cameroun, du Nigeria et du Tchad, et les conditions climatiques et socio-démographiques sont semblables dans les régions frontalières de ces trois pays. Les cPVDV originaires du Cameroun, du Tchad et du Nigéria, qui ont été caractérisés, se sont avérés être des recombinants entre des souches du VPO et d’autres HEV-C non vaccinaux [26]. Ces résultats sont donc en accord avec l’hypothèse selon laquelle l’intense circulation de divers HEV-C dans des pays où la couverture vaccinale est insuffisante serait un facteur viral favorisant l’émergence de cPVDV recombinants pathogènes.
Quel futur pour l’éradication ?
L’éradication de la PPA due au PV reste un objectif qu’il est possible d’atteindre, et certains succès sont très encourageants, comme la disparition des souches sauvages de PV de type 2, la raréfaction des souches de type 3, et la récente élimination de tous les PV sauvages de l’Inde [2]. L’éradication mondiale de toutes les souches de PV sauvages est néanmoins plus difficile que prévu en 1988 [28], et requiert des fonds supplémentaires. La collaboration entre les chercheurs de différents instituts du réseau international des Instituts Pasteur a permis de révéler l’émergence à Madagascar de cPVDV, virus neuropathogènes capables de se propager chez l’homme de manière silencieuse et de causer des épidémies de PPA [27]. Ces épidémies peuvent heureusement être prévenues ou stoppées par des campagnes vaccinales efficaces, mais la prévention requiert aussi une surveillance par des laboratoires compétents, capables d’utiliser une technologie de pointe. En 2011 à Madagascar, c’est l’association de ces compétences qui a permis de mettre en évidence la ré-émergence de cPVDV, d’alerter les autorités et, très probablement, de prévenir de nouvelles épidémies par une riposte vaccinale adaptée [17]. Il faut aussi souligner l’importance de la recherche fondamentale visant à comprendre les mécanismes d’émergence de ces virus. Là encore, les études menées à Madagascar, au Cameroun, et dans les autres pays du REIP, en étroite collaboration avec l’Institut Pasteur de Paris, ont montré la parenté entre certaines séquences du génome des cPVDV et celles de souches locales de CVA de la même espèce, en particulier les CVA-17 et CVA-13, suggérant fortement un rôle de la recombinaison entre PV et CVA de la même espèce, dans l’émergence des cPVDV [4, 20]. Ces travaux de recherche, ainsi que ceux d’autres collègues à l’étranger, ont déjà des conséquences pour l’amélioration des stratégies de vaccination et de surveillance. Santé publique et surveillance sont des aspects appliqués de la recherche fondamentale qui doit rester dynamique jusqu’à la disparition, non seulement des souches sauvages, mais aussi des souches vaccinales et dérivées du vaccin.
Il semble probable que l’éradication de la PPA se fasse en deux phases successives : la première ciblant préférentiellement les souches sauvages et la seconde concernant plutôt les souches dérivées du vaccin. Au cours de la première phase, un VPO bivalent (type 1-type 3) est parfois utilisé localement parce que ce vaccin induit une meilleure réponse contre les types 1 et 3 que le VPO trivalent, et que les souches sauvages de type 2 ont disparu. Cependant, le danger d’émergence de cPVDV constitue la raison essentielle qui pousse à abandonner l’emploi du VPO dès que possible. Aussi, dans une seconde phase, après l’éradication de toutes les souches sauvages, le VPO devrait être remplacé par le VPI, pendant une période qui reste à déterminer. Ce processus est d’ores et déjà envisagé en ce qui concerne les souches de PV de type 2.
D’autres vaccins sont aussi à l’étude. Des interventions génétiques visent à freiner la vitesse d’évolution des souches du VPO. Il est ainsi possible de rendre la polymérase virale plus fidèle, et donc moins mutagène lors de la réplication du virus, ce qui stabilise le phénotype viral. Des études récentes ont aussi montré que l’on pouvait reprogrammer le génome du PV en remplaçant un grand nombre de codons par des homologues synonymes, sans changer la séquence des protéines virales, ce qui permettait de créer des souches très atténuées. Par ailleurs, certaines manipulations tendent à diminuer la virulence des souches utilisées pour la préparation du VPI. Il est par exemple possible de remplacer dans le génome du PV, le site interne d’initiation de la traduction par celui d’un rhinovirus, agent du rhume génétiquement apparenté au PV. Le PV recombinant perd alors sa capacité à se multiplier dans le système nerveux central, et perd donc sa neurovirulence, sans perdre sa capacité à se multiplier dans des cellules non nerveuses. Utiliser des souches non pathogènes de PV pour la préparation du VPI permettrait de restreindre les conditions de sécurité et de confinement pour les amplifier, donc de diminuer considérablement les coûts du VPI. Cela rendrait également sa fabrication plus accessible à des pays en voie de développement. L’ensemble de ces recherches sont autant de pistes pour l’élaboration de nouveaux vaccins plus sûrs, qu’il s’agisse du PV ou d’autres virus.
Après l’éradication, il existera encore un risque, même s’il est faible, de réémergence de la poliomyélite à partir de souches présentes, par exemple, dans des échantillons biologiques de laboratoires. Un autre danger pourrait venir des virus excrétés par des personnes immuno-déficientes qui peuvent être chroniquement infectées pendant des mois, voire des années, par des souches de PV pathogènes d’origine vaccinale. L’hypothèse d’une réémergence du PV à partir des CVA les plus proches génétiquement a été formulée, et ne peut être complètement écartée. Pour toutes ces raisons, la vaccination antipoliomyélitique, accompagnée d’une surveillance rigoureuse, devra donc être poursuivie pendant de nombreuses années, avant que l’on puisse s’affranchir de tout risque de réémergence et certifier l’éradication de la poliomyélite et des PV.
Remerciements
Nous remercions pour leur soutien, la Division internationale de l’Institut Pasteur, l’Inserm, la Société de pathologie exotique, l’Agence nationale pour la recherche (ANR 09 MIEN 019), la Fondation pour la recherche médicale (FRM DMI20091117313), le ministère français des Affaires Étrangères et Européennes et l’Organisation mondiale de la santé (OMS HQPOL1206310).
Consulter http://www.polioeradication.org.
Consulter http://www.polioeradication.org/Dataandmonitoring.
Références
- Guerin N, Delpeyroux F, Rey M. Poliomyélite. EMC, Maladies infectieuses. Paris : Elsevier Masson, 2007. [Google Scholar]
- Anonymous. Update on vaccine-derived polioviruses-worldwide, april 2011-june 2012. MMWR Morb Mortal Wkly Rep 2012 ; 61 : 741–746. [PubMed] [Google Scholar]
- Pallansch MA, Roos R, Enteroviruses : polioviruses, coxsackieviruses, echoviruses, and newer enteroviruses. In : Knipe DM, Howley PM, eds. Fields Virology. Philadelphia : Lippincott Williams and Wilkins, 2007. [Google Scholar]
- Blondel B, Autret A, Brisac C, et al. Genetic evolution of poliovirus : success and difficulties in the eradication of paralytic poliomyelitis. Med Trop 2008 ; 68 : 189–202. [Google Scholar]
- Strebel PM, Aubert-Combiescu A, Ion-Nedelcu N, et al. Paralytic poliomyelitis in Romania, 1984–1992. Evidence for a high risk of vaccine-associated disease and reintroduction of wild-virus infection. Am J Epidemiol 1994 ; 140 : 1111–1124. [PubMed] [Google Scholar]
- Kew OM, Sutter RW, de Gourville EM, et al. Vaccine-derived polioviruses and the endgame strategy for global polio eradication. Annu Rev Microbiol 2005 ; 59 : 587–635. [CrossRef] [PubMed] [Google Scholar]
- Kew O, Morris-Glasgow V, Landaverde M, et al. Outbreak of poliomyelitis in Hispaniola associated with circulating type 1 vaccine-derived poliovirus. Science 2002 ; 296 : 356–359. [CrossRef] [PubMed] [Google Scholar]
- Shimizu H, Thorley B, Paladin FJ, et al. Circulation of type 1 vaccine-derived poliovirus in the Philippines in 2001. J Virol 2004 ; 78 : 13512–13521. [CrossRef] [PubMed] [Google Scholar]
- Yang CF, Naguib T, Yang SJ, et al. Circulation of endemic type 2 vaccine-derived poliovirus in Egypt from 1983 to 1993. J Virol 2003 ; 77 : 8366–8377. [CrossRef] [PubMed] [Google Scholar]
- Romanenkova NI, Guillot S, Rozaeva NR, et al. Use of a multiple restriction fragment length polymorphism method for detecting vaccine-derived polioviruses in clinical samples. J Clin Microbiol 2006 ; 44 : 4077–4084. [CrossRef] [PubMed] [Google Scholar]
- Rousset D, Rakoto-Andrianarivelo M, Razafindratsimandresy R, et al. Poliovirus vaccination and emergence of recombinant vaccine derived from vaccinal strains in Madagascar. Med Malad Infect 2004 ; 34 : S65–S66. [CrossRef] [Google Scholar]
- Rousset D, Rakoto-Andrianarivelo M, Razafindratsimandresy R, et al. Recombinant vaccine-derived poliovirus in Madagascar. Emerg Infect Dis 2003 ; 9 : 885–887. [CrossRef] [PubMed] [Google Scholar]
- Rakoto-Andrianarivelo M, Guillot S, Iber J, et al. Co-circulation and evolution of polioviruses and species C enteroviruses in a district of Madagascar. PLoS Pathog 2007 ; 3 : e191. [CrossRef] [PubMed] [Google Scholar]
- Rakoto-Andrianarivelo M, Gumede N, Jegouic S, et al. Reemergence of recombinant vaccine-derived poliovirus outbreak in Madagascar. J Infect Dis 2008 ; 197 : 1427–1435. [CrossRef] [PubMed] [Google Scholar]
- Joffret ML, Jegouic S, Bessaud M, et al. Common and diverse features of cocirculating type 2 and 3 recombinant vaccine-derived polioviruses isolated from patients with poliomyelitis and healthy children. J Infect Dis 2012 ; 205 : 1363–1373. [CrossRef] [PubMed] [Google Scholar]
- Bessaud M, Joffret ML, Holmblat B, et al. Genetic relationship between cocirculating human enteroviruses of species C. PLoS One 2011 ; 6 : e24823. [CrossRef] [PubMed] [Google Scholar]
- Razafindratsimandresy R, Joffret ML, Rabemanantsoa S, et al. Re-emergence of recombinant vaccine-derived polioviruses in healthy children in Madagascar. Emerg Infect Dis 2013 ; 19 : 1008–1010. [CrossRef] [PubMed] [Google Scholar]
- Jegouic S, Joffret ML, Blanchard C, et al. Recombination between polioviruses and co-circulating Coxsackie A viruses : role in the emergence of pathogenic vaccine-derived polioviruses. PLoS Pathog 2009 ; 5 : e1000412. [CrossRef] [PubMed] [Google Scholar]
- Riquet FB, Blanchard C, Jegouic S, et al. Impact of exogenous sequences on the characteristics of an epidemic type 2 recombinant vaccine-derived poliovirus. J Virol 2008 ; 82 : 8927–8932. [CrossRef] [PubMed] [Google Scholar]
- Combelas N, Holmblat B, Joffret ML, et al. Recombination between poliovirus and coxsackie A virus of species C: a model of viral genetic plasticity and emergence. Viruses 2011 ; 3 : 1460–1484. [CrossRef] [PubMed] [Google Scholar]
- Wassilak S, Pate MA, Wannemuehler K, et al. Outbreak of type 2 vaccine-derived poliovirus in Nigeria : emergence and widespread circulation in an underimmunized population. J Infect Dis 2011 ; 203 : 898–909. [CrossRef] [PubMed] [Google Scholar]
- Baicus A, Persu A, Dinu S, et al. The frequency and biodiversity of poliovirus and non-polio enterovirus strains isolated from healthy children living in a limited area in Romania. Arch Virol 2011 ; 156 : 701–706. [CrossRef] [PubMed] [Google Scholar]
- Haddad-Boubaker S, Yahia AB, Rezig D, et al. Enterovirus detection in stool specimen: relevance for poliovirus and enterovirus surveillance. Arch Inst Pasteur Tunis 2007 ; 84 : 3–9. [PubMed] [Google Scholar]
- Bessaud M, Pillet S, Ibrahim W, et al. Molecular characterization of human enteroviruses in the Central African Republic: uncovering wide diversity and identification of a new human enterovirus A71 genogroup. J Clin Microbiol 2012 ; 50 : 1650–1658. [CrossRef] [PubMed] [Google Scholar]
- Sadeuh-Mba SA, Bessaud M, Massenet D, et al. High frequency and diversity of species C enteroviruses in Cameroon and neighboring countries. J Clin Microbiol 2013 ; 51 : 759–770. [CrossRef] [PubMed] [Google Scholar]
- Burns CC, Shaw J, Jorba J, et al. Multiple independent emergences of type 2 vaccine-derived polioviruses during a large outbreak in northern Nigeria. J Virol 2013 ; 87 : 4907–4922. [CrossRef] [PubMed] [Google Scholar]
- Rakoto-Andrianarivelo M, Jegouic S, Bessaud M, Delpeyroux F. Poliovirus et entérovirus C, même espèce, même tribu virale. Med Sci (Paris) 2008 ; 24 : 452–453. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Antona D. L’éradication des maladies infectieuses : l’exemple de la poliomyélite. Med Sci (Paris) 2002 ; 18 : 55–61. [CrossRef] [EDP Sciences] [Google Scholar]
Liste des figures
|
Figure 1. Localisation des cas de poliomyélite dus à des cPVDV de type 2 ou 3 à Madagascar en 2001, 2002 et 2005. Chaque point représente un cas de poliomyélite et probablement 100 à 1 000 personnes infectées mais dépourvues de symptômes. Le nom du district et l’année où le cas a été rapporté sont indiqués. La localisation de la capitale du Pays, Antananarivo, est également mentionnée. |
| Dans le texte | |
|
Figure 2. Structure schématique des génomes de différents cPVDV recombinants isolés à Madagascar de 2001 à 2011. L’organisation génomique du poliovirus indique les différentes régions 5’ et 3’ non codantes (5’ NC et 3’ NC), ainsi que la phase ouverte de lecture codant pour les protéines structurales (en bleu) et non structurales (en jaune et en vert). La longueur du génome est indiquée (une unité = 103 nucléotides). L’origine des séquences constituant les génomes recombinants est indiquée selon qu’elle se réfère à des séquences d’origine vaccinale (Sabin) mutées ou à des séquences d’autres HEV-C. Les séquences se rapprochant de celles de souches de virus coxsackie connues sont indiquées. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.