")
")
| Issue |
Med Sci (Paris)
Volume 29, Number 3, Mars 2013
|
|
|---|---|---|
| Page(s) | 245 - 247 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/2013293005 | |
| Published online | 27 mars 2013 | |
Rôle de l’hormone thyroïdienne T3 dans la régénération axonale chez les vertébrés supérieurs
Thyroid hormone T3 triggers the developmental loss of axonal regenerative capacity
1
Neurobiologie des processus adaptatifs, UMR7102, CNRS, UPMC, 9, quai Saint Bernard, 75005 Paris, France
2
Institut de génomique fonctionnelle de Lyon, ENS de Lyon, CNRS, Inra, Université de Lyon, Lyon, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Après une lésion, les neurones des mammifères adultes ne régénèrent pas spontanément leur axone dans le système nerveux central (SNC, comprenant le cerveau et la moelle épinière). Cette incapacité contribue aux déficits fonctionnels importants que créent chez les patients les lésions traumatiques, notamment de la moelle épinière. La plupart des tentatives destinées à promouvoir la régénération axonale dans le SNC des mammifères adultes visent à modifier l’environnement des axones sectionnés [1]. Le succès mitigé de ces tentatives suggère qu’au cours de leur développement, la plupart des neurones répriment leur programme de croissance axonale de manière définitive et deviennent incapables de régénérer leur axone, même s’ils sont placés dans un environnement permissif pour la croissance axonale [2].
Implication de l’hormone thyroïdienne T3 dans la perte de la capacité de régénération axonale
Les mécanismes sous-jacents à cette perte de capacité de croissance axonale sont encore mal connus. Beaucoup de travaux suggèrent que la myélinisation, qui n’intervient qu’après la pousse de l’axone, est responsable de ce processus [1]. Toutefois, il semble que pour certains neurones, des facteurs de transcription régulent de manière autonome la perte de capacité de régénération axonale. C’est le cas notamment des neurones ganglionnaires de la rétine [3].
Pour de nombreux types de neurones de souris (par exemple les neurones du cortex enthorinal, les cellules ganglionnaires de la rétine et les cellules de Purkinje), la capacité de régénération axonale disparaît au cours de la première semaine de vie postnatale [2]. Or, à ce stade de développement, on observe une augmentation temporaire du niveau de l’hormone thyroïdienne T3 dans la circulation sanguine. De façon intéressante, chez les amphibiens comme le xénope, la perte de capacité régénérative des neurones se produit juste après la métamorphose [4], qui est elle-même déclenchée par la T3. Dans le cadre d’un travail récemment publié dans les Proceedings of the National Academy of Sciences of USA, nous avons cherché à savoir si la T3 pouvait être impliquée dans la perte des capacités régénératives axonales au cours du développement d’un type de neurones, les cellules de Purkinje du cervelet [5].
Nous avons utilisé un modèle de culture organotypique, permettant d’isoler une région cérébrale tout en conservant une grande partie de la structure du tissu et les interactions cellulaires qui s’y produisent. Ceci nous a permis de montrer qu’en présence d’un excès de T3, les cellules de Purkinje de souris perdent prématurément la capacité de régénérer leur axone. A contrario, chez des animaux rendus hypothyroïdiens par l’administration d’une molécule éliminant la T3 sanguine, ces neurones perdent plus tardivement cette capacité (Figure 1).
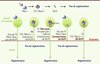 |
Figure 1. L’hormone thyroïdienne (T3) inhibe la régénération axonale via sa liaison à son récepteur TRα1 et à l’induction de l’expression du facteur de transcription Klf9. L’utilisation de souris transgéniques ou de vecteurs lentiviraux nous a permis de montrer l’implication de T3 et TRα1, et de l’expression du facteur de transcription Klf9 dans la perte des capacités de la cellule de Purkinje à régénérer son axone. |
Rôle des récepteurs de la T3 et du facteur de transcription KLF9 dans la régénération axonale
La T3 agit sur la transcription génique en se liant à des récepteurs nucléaires, TRα1 et TRβ1/2. Ces récepteurs ont des propriétés proches. Ils sont fixés à l’ADN par leur partie amino-terminale, et activent la transcription de nombreux gènes cibles quand l’hormone vient se fixer dans la partie carboxy-terminale. Ainsi, par l’utilisation d’une souris mutante chez laquelle TRα1 a perdu sa capacité d’activer la transcription, nous avons pu montrer que ce récepteur régule l’expression de gènes impliqués dans la perte de la capacité régénérative des cellules de Purkinje (Figure 1). Au cours du développement postnatal de la souris, le pic physiologique d’augmentation de T3 détermine donc le moment auquel les cellules de Purkinje perdent leur capacité à régénérer leur axone.
Par ailleurs, nous avons montré que la T3 induisait l’expression du facteur de transcription Krüppel-like factor 9 (KLF9) dans les cellules de Purkinje. En utilisant des approches de gain et de perte de fonction, nous avons également démontré l’implication de KLF9 dans la perte de capacité des cellules de Purkinje à régénérer leur axone, suggérant que ce facteur de transcription représente un maillon essentiel du processus conduisant à la perte de la capacité de régénération axonale dans ces cellules (Figure 1). De façon remarquable, ce facteur est également impliqué dans la perte de capacité de régénération axonale des cellules ganglionnaires de la rétine [3], ce qui suggère une probable similitude des mécanismes moléculaires assurant cette régulation dans différentes populations neuronales.
La T3 : une responsabilité dans l’autonomisation des individus ?
Le rôle de la T3 dans la régénération axonale a été bien démontré chez le xénope [4], et nous venons de le démontrer chez la souris pour un type de neurone, la cellule de Purkinje [5]. Mais le rôle de la T3 dans les cellules nerveuses ne se limite pas à cette fonction, car l’hormone participe, notamment, au déclenchement de la myélinisation [6]. De façon plus générale, l’ensemble des changements morphologiques et électrophysiologiques que subit la cellule de Purkinje au moment du pic de T3 sont tellement importants qu’il est possible de les comparer à une « métamorphose cellulaire » [7], comme cela a été précédemment proposé pour les cellules de l’intestin [8]. Nous avons donc analysé dans la littérature si la perte des capacités régénératives pouvait faire partie d’un programme plus vaste de maturation neuronale, qui serait conservé au cours de l’évolution et déclenché par la T3.
Cette analyse nous conduit à proposer que la T3 soit responsable de l’autonomisation des individus. En effet, les oiseaux et les mammifères peuvent être divisés en espèce nidifuges - animaux vivants hors d’un nid dès l’éclosion ou la naissance (poulet, mouton) -, et en espèces nidicoles - vivant dans un nid à l’éclosion ou à la naissance (pigeon, souris). Chez les animaux nidifuges (Figure 2), la fonction thyroïdienne se développe pendant la période prénatale. En conséquence, les systèmes moteurs et sensoriels sont déjà fonctionnels à la naissance [9, 10], et l’animal se déplace de façon autonome. De manière remarquable, la perte des capacités régénératives axonales se produit également pendant la période prénatale chez le poulet et le mouton [11, 12]. A contrario, chez les animaux nidicoles, la maturation de la thyroïde, tout comme celle des systèmes moteurs et sensoriels, se produit après la naissance [9], coïncidant également avec la perte des capacités de régénération axonale. Il semble donc qu’il y ait concordance entre ces deux aspects du neurodéveloppement. Une des modulations possible de ce programme de maturation cérébrale découle de la régulation autonome du taux de T3 cérébrale. On sait, en effet, que le taux intracérébral de T3 peut différer notablement de son taux sérique. Ceci résulte de la capacité des cellules gliales d’assurer une synthèse locale de T3 à partir de la thyroxine présente dans le sang, et de la capacité des neurones à dégrader la T3 [13].
 |
Figure 2. Hypothèse d’un vaste programme de maturation cérébrale orchestré par l’hormone thyroïdienne T3. La T3 synchronise les adaptations qui sont nécessaires à un organisme juvénile pour se mouvoir indépendamment dans son milieu naturel. La perte de la capacité des neurones à régénérer leur axone dans le système nerveux central ferait partie de ce programme. Les espèces nidicoles ont un pic de T3 après leur naissance et perdent leur capacité à régénérer ensuite, alors que les espèces nidifuges perdent ces capacités dans l’œuf ou pendant la gestation, parallèlement au pic de T3. |
Le cas de l’homme peut apparaître comme une exception à la règle qui semble émerger de notre analyse associant la T3 à un programme de maturation cérébrale nécessaire à l’acquisition de l’autonomie. En effet la première augmentation d’hormone thyroïdienne circulante se produit chez l’homme pendant la période embryonnaire, alors que les fonctions sensorielles et motrices n’apparaissent pas avant la naissance. Toutefois, le développement chez l’homme est extrêmement ralenti : la myélinisation, par exemple, commence dès la période embryonnaire, puis se poursuit tout au long des premières années de la vie. Il est ainsi possible que, comme la plupart des primates, l’homme soit, à l’origine, une espèce nidifuge. Elle aurait subi, ensuite, une évolution dite néoténique [14], tendant à prolonger les stades juvéniles de développement et retardant l’apparition de l’autonomie.
Conclusion
Ainsi, l’ensemble de ces données nous permettent de proposer que la perte des capacités de régénération axonale dans le SNC des mammifères supérieurs fait partie d’un programme plus vaste, permettant de synchroniser les adaptations qui sont nécessaires à un organisme juvénile pour se mouvoir indépendamment, et évoluer dans son milieu naturel. En ce sens, ce vaste programme pourrait être considéré comme une réminiscence de la métamorphose observée chez les amphibiens. Il reste cependant à découvrir les composants de cette métamorphose effectivement déclenchés par la T3, et à comprendre l’avantage sélectif que procure cette réduction considérable des capacités de réparation et de plasticité neuronales au cours de l’évolution des vertébrés. Cette analyse apportera peut-être des idées nouvelles pour traiter les lésions aujourd’hui irréversibles du système nerveux central.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Références
- Schwab ME. Nogo and axon regeneration. Curr Opin Neurobiol 2004 ; 14 : 118–124. [CrossRef] [PubMed] [Google Scholar]
- Goldberg JL. Intrinsic neuronal regulation of axon and dendrite growth. Curr Opin Neurobiol 2004 ; 14 : 551–555. [CrossRef] [PubMed] [Google Scholar]
- Moore D, Blackmore M, Hu Y, et al. KLF family members regulate intrinsic axon regeneration ability. Science 2009 ; 326 : 298. [CrossRef] [PubMed] [Google Scholar]
- Beattie MS, Bresnahan JC, Lopate G. Metamorphosis alters the response to spinal cord transection in Xenopus laevis frogs. J Neurobiol 1990 ; 21 : 1108–1122. [CrossRef] [PubMed] [Google Scholar]
- Avci HX, Lebrun C, Wehrle R, et al. Thyroid hormone triggers the developmental loss of axonal regenerative capacity via thyroid hormone receptor α1 and kruppel-like factor 9 in Purkinje cells. Proc Natl Acad Sci USA 2012 ; 109 : 14206–14211. [CrossRef] [Google Scholar]
- Oppenheimer JH, Schwartz HL. Molecular basis of thyroid hormone-dependent brain development. Endocr Rev 1997 ; 18 : 462–475. [CrossRef] [PubMed] [Google Scholar]
- Dusart I, Flamant F. Profound morphological, functional changes of rodent Purkinje cells between the first, the second postnatal weeks: a metamorphosis ? Front Neuroanat 2012 ; 6 : 11. [CrossRef] [PubMed] [Google Scholar]
- Kress E, Samarut J, Plateroti M. Thyroid hormones and the control of cell proliferation or cell differentiation: paradox or duality ? Mol Cell Endocrinol 2009 ; 313 : 36–49. [CrossRef] [PubMed] [Google Scholar]
- McNabb FMA. Avian thyroid development and adaptive plasticity. Gen Comp Endocrinol 2006 ; 147 : 93–101. [CrossRef] [PubMed] [Google Scholar]
- Fisher DA, Polk DH, Wu SY. Fetal thyroid metabolism: a pluralistic system. Thyroid 1994 ; 4 : 367–371. [CrossRef] [PubMed] [Google Scholar]
- Hasan SJ, Keirstead HS, Muir GD, Steeves JD. Axonal regeneration contributes to repair of injured brainstem-spinal neurons in embryonic chick. J Neurosci 1993 ; 13 : 492–507. [PubMed] [Google Scholar]
- Meuli-Simmen C, Meuli M, Hutchins GM, et al. The fetal spinal cord does not regenerate after in utero transection in a large mammalian model. Neurosurgery 1996 ; 39 : 555–561. [PubMed] [Google Scholar]
- St Germain DL, Galton VA, Hernandez A. Defining the roles of the iodothyronine deiodinases: current concepts and challenges. Endocrinology 2008 ; 150 : 1097–1107. [CrossRef] [Google Scholar]
- Gould SJ. Le véritable père de l’homme est l’enfant. In : Darwin et les grandes énigmes de la vie. Paris : Seuil, 1984 : 64–71. [Google Scholar]
© 2013 médecine/sciences – Inserm / SRMS
Liste des figures
|
Figure 1. L’hormone thyroïdienne (T3) inhibe la régénération axonale via sa liaison à son récepteur TRα1 et à l’induction de l’expression du facteur de transcription Klf9. L’utilisation de souris transgéniques ou de vecteurs lentiviraux nous a permis de montrer l’implication de T3 et TRα1, et de l’expression du facteur de transcription Klf9 dans la perte des capacités de la cellule de Purkinje à régénérer son axone. |
| Dans le texte | |
|
Figure 2. Hypothèse d’un vaste programme de maturation cérébrale orchestré par l’hormone thyroïdienne T3. La T3 synchronise les adaptations qui sont nécessaires à un organisme juvénile pour se mouvoir indépendamment dans son milieu naturel. La perte de la capacité des neurones à régénérer leur axone dans le système nerveux central ferait partie de ce programme. Les espèces nidicoles ont un pic de T3 après leur naissance et perdent leur capacité à régénérer ensuite, alors que les espèces nidifuges perdent ces capacités dans l’œuf ou pendant la gestation, parallèlement au pic de T3. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.