")
")
| Issue |
Med Sci (Paris)
Volume 29, Number 1, Janvier 2013
|
|
|---|---|---|
| Page(s) | 83 - 88 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/2013291017 | |
| Published online | 25 janvier 2013 | |
Les cancers des voies aérodigestives supérieures associés aux papillomavirus
Cancers of the upper aerodigestive tract associated with human papillomavirus
1
Service d’anatomie pathologique, hôpital européen Georges Pompidou, 20-40, rue Leblanc, 75015 Paris, France
2
Inserm U970, équipe 10, faculté Paris-Descartes, Paris-centre de recherche cardiovasculaire (PARCC), 56, rue Leblanc, 75015 Paris, France
3
Laboratoire de virologie, hôpital européen Georges Pompidou, 20-40, rue Leblanc, 75015 Paris, France
4
Laboratoire d’immunologie, hôpital européen Georges Pompidou, 20-40, rue Leblanc, 75015 Paris, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Les carcinomes des voies aérodigestives sont le plus souvent secondaires à une intoxication alcoolo-tabagique. Cependant, il a été démontré que les papillomavirus oncogènes humains (HPV) ont un rôle de plus en plus important dans la genèse de ces cancers. Les carcinomes HPV+ sont le plus souvent observés chez des sujets plus jeunes que ceux porteurs de carcinomes secondaires à une intoxication alcoolo-tabagique. Ils sont principalement localisés au niveau de l’oropharynx et, en particulier, au niveau de l’amygdale. Le virus HPV est détecté dans les cancers de l’oropharynx avec une fréquence variant de 40 à 90 % suivant l’origine géographique des patients. Ces carcinomes HPV+ sont de meilleur pronostic et leur radio- ou chimiosensibilité est meilleure. Pour l’instant, aucune modification de traitement n’est recommandée, mais plusieurs essais thérapeutiques sont en cours. La vaccination préventive des garçons devient une véritable question de santé publique, ce d’autant qu’elle est déjà en place dans certains pays. D’autre part, une meilleure compréhension du microenvironnement tumoral de ces cancers permettra, à terme, de proposer une vaccination thérapeutique.
Abstract
Carcinomas of the aerodigestive tract are most often secondary to alcohol and tobacco intoxication. However, it is shown that the oncogenic human papillomavirus (HPV) have an increasing role in the carcinogenesis of these cancers. Patients with HPV+ carcinoma are generally younger and not alcohol and tobacco users. These carcinomas are mainly localized in the oropharynx and in particular at the tonsil. HPV is found in 40 to 90 % of the cancers in the oropharynx, depending on the country. These HPV+ carcinomas have a better prognosis with better radio or chemosensitivity. To date, no change of treatment is recommended, however, several trials are underway. Preventive vaccination of boys is a real public health issue, especially since it is recommended in some countries. Moreover, a better understanding of the tumor microenvironment will ultimately offer therapeutic vaccination.
© 2013 médecine/sciences – Inserm / SRMS
Plus de 80 % des tumeurs malignes des voies aérodigestives supérieures sont des carcinomes épidermoïdes. Ces cancers sont de mauvais pronostic et la survie des patients à 5 ans n’est que d’environ 50 % [1], malgré les avancées thérapeutiques comportant de la radiothérapie et/ou de la chimiothérapie, associée ou non à une chirurgie et à des thérapies ciblées (cetuximab, ou erbitux® dirigé contre l’EGF-R). Les carcinomes épidermoïdes des voies aérodigestives supérieures (CEVADS) sont majoritairement secondaires à une intoxication alcoolo-tabagique marquée. Depuis les années 1980, l’implication de certains papillomavirus humains (HPV) dans la genèse de ces cancers a été suspectée [2], puis démontrée. L’incidence de l’infection par HPV dans le monde est très forte puisque plus de 660 millions de personnes sont infectées. La responsabilité de la famille des HPV est incriminée dans environ 5 % de la totalité des cancers (cancers du col de l’utérus, de la vulve, du vagin, de l’anus). Pour ce qui est des CEVADS, son implication n’est plus discutée, et il apparaît que ce virus est le facteur de risque principal dans certaines localisations dont l’oropharynx. D’autre part, il semble que les cancers des VADS induits par les virus soient de meilleur pronostic que ceux qui sont secondaires à un alcoolo-tabagisme. Dans cette revue, nous présentons le rôle de l’HPV dans la cancérogenèse et les spécificités de ces carcinomes induits par les virus.
HPV et cancers des VADS : épidémiologie
Les CEVADS occupent la 5e place en termes de fréquence des cancers dans le monde. Les principaux facteurs de risque sont l’exposition au tabac et à l’alcool. La baisse de consommation de ces produits explique la diminution du nombre de cancers du larynx. Cependant, dans d’autres localisations, et, en particulier, au niveau de l’oropharynx (amygdales et base de la langue), l’incidence de ces cancers ne cesse d’augmenter depuis plusieurs années [3, 4]. Les facteurs de risque habituels, ou autres (marijuana, bétel), n’expliquent pas cette évolution épidémiologique. K. Syrjänen fut la première à suspecter l’implication de certains HPV oncogènes dans la genèse des CEVADS [2, 5]. Une étude américaine montre une augmentation de l’incidence annuelle des CEVADS HPV+ de 0,65 %, alors que l’incidence de ceux qui sont liés à l’alcool et au tabac baisse de 2,42 % chaque année depuis 1983 [6]. L’implication de l’HPV semble varier en fonction du site lésionnel et de l’origine géographique des patients. Une méta-analyse mondiale de 5 046 patients estime la prévalence des HPV dans ces CEVADS à environ 26 % des cas, toutes localisations confondues [7]. De façon générale, la présence de virus HPV oncogènes est trouvée dans environ 36 % des cancers de l’oropharynx (base de la langue et amygdale) [8]. Dans certaines régions, comme en Europe du Nord ou en Amérique du Nord, cette proportion peut atteindre plus de 80 % ; elle est de 46,5 % en France [9]. Pour certains, cette incidence croissante a des allures d’épidémie ou de pandémie virale [3, 10].
Le génotype HPV16 est majoritaire dans les CEVADS, suivi du génotype HPV18. Quant aux génotypes HPV31, 33, 35, 45, 51, 52, 56, 58, 59, 68 et 82, ils ont été trouvés au moins une fois [7, 11].
Le mode de transmission de l’HPV au niveau des VADS est principalement lié à une activité sexuelle et à ses pratiques (nombre de partenaires, âge des premiers rapports, type de relation, etc.) [12, 13]. Cependant, d’autre modes de transmission sont suspectés, comme le suggère la détection (PCR) de HPV oncogènes dans des pièces d’amygdalectomie chez les enfants [14, 15].
Structure des papillomavirus
Les papillomavirus sont de petits virus nus, de 45 à 55 nm de diamètre, non enveloppés, dont la capside à symétrie icosaédrique est constituée de 72 capsomères. Le génome viral est constitué d’une molécule d’ADN bicaténaire et circulaire d’environ 8 000 paires de bases. Un seul des deux brins d’ADN est codant avec des phases ouvertes de lecture qui se chevauchent. Ce génome est organisé en trois régions : la région régulatrice non codante LCR (locus control region), la région E (early) et la région L (late) (Figure 1). Plus de 120 génotypes de HPV différents ont été décrits, dont une quarantaine détectés au niveau de la sphère anogénitale. La récente classification proposée par l’Agence internationale de recherche sur le cancer (IARC) a permis de classer les HPV génitaux selon le risque oncogène. Cette nouvelle classification, qui s’appuie sur plusieurs études internationales, fait maintenant référence. Elle distingue : (1) le groupe des HPV à haut risque (HPV-HR) : 16, 18, 26, 31, 33, 35, 39, 45, 51, 52, 53, 56, 58, 59, 66, 68, 73 et 82 ; (2) le groupe des HPV à bas risque (HPV-BR) : 6, 11, 40, 42, 43, 44, 54, 61, 70, 72, 81 et CP6108 ; et (3) le groupe des HPV à risque inconnu (HPV-RI) qui rassemble tous les génotypes n’appartenant pas aux deux groupes précédents.
 |
Figure 1. Représentation du génome HPV-16. Les phases ouvertes de lecture sont nommées E1, E2, E4, E5, E6 et E7 pour les protéines précoces (E pour early) et L1 et L2 pour les protéines tardives (L pour late). La région LCR (locus control region) contient les éléments de régulation de la transcription et de la régulation. |
HPV et cancers des VADS : mécanismes oncogéniques
Le mécanisme oncogénique central est porté par les protéines virales E6 et E7 et dépend de l’intégration du génome HPV à haut risque au génome de l’hôte. La protéine virale E7 se lie à la protéine du rétinoblastome (pRB) et conduit à sa dégradation par le protéasome. Ceci peut aboutir à la dérégulation du cycle cellulaire (induction de la progression en phase S par la libération de la protéine E2F du complexe pRb/E2F) et à la transformation cellulaire (voir plus loin et Figure 2). La protéine E6, quant à elle, se lie à p53 qui est un facteur proapoptotique. Une fois dégradée, p53 ne peut plus exercer son rôle de « garde-fou » ce qui peut conduire à l’immortalisation de ces cellules. En cas d’intégration du génome HPV, la région E2 est clivée ce qui entraîne une levée de l’inhibition exercée par la protéine virale E2 sur l’expression des protéines E6 et E7. La surexpression de ces deux protéines oncogéniques conduit à la dérégulation du cycle cellulaire, et donc à l’immortalisation et à la transformation de la cellule épithéliale. Ces deux protéines sont essentielles aux propriétés oncogéniques du virus HPV, mais ne sont pourtant pas suffisantes. La protéine E5 semble aussi jouer un rôle dans le processus de transformation tumorale. Cette protéine est capable d’induire la transformation des fibroblastes et des kératinocytes de rongeurs, et augmente le pouvoir transformant de certains oncogènes comme Ras.
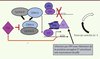 |
Figure 2. Interférence de la protéine virale E7 avec le point de contrôle G1-S de la mitose. pRb se lie au facteur de transcription E2F et provoque son inhibition, empêchant ainsi l’expression de la cycline E et une série de gènes indispensables à la phase S. L’oncoprotéine virale E7, produite par les virus oncogènes HPV, présente une forte affinité pour pRb et se lie particulièrement à la forme hypophosphorylée (la forme active). La dissociation du complexe pRB-E2F libère E2F sous sa forme active, ce qui permet le déblocage du cycle cellulaire et entraîne la surexpression de p16, qui est une protéine de rétrocontrôle. |
HPV et cancers des VADS : aspects histologiques et mode de détection de l’HPV
Les carcinomes épidermoïdes induits par l’HPV peuvent correspondre à plusieurs sous-types histologiques. Cependant, au niveau de l’oropharynx, et en particulier de l’amygdale, on observe de façon assez récurrente un sous-type histologique particulier : le carcinome épidermoïde basaloïde. Ce carcinome peu différencié se caractérise par la présence de massifs constitués de petites cellules basales, avec une zone de nécrose centrale (comédonécrose). Au niveau de l’amygdale, ce carcinome est le plus souvent associé à une infection par HPV et ne présente pas les mêmes caractéristiques évolutives que les carcinomes peu différenciés, car il semble être plutôt de bon pronostic [16, 17]. Il existe le plus souvent une inactivation des protéines p53 et pRb dans les CEVADS, indépendamment du statut HPV. Dans les cancers HPV+, la protéine p53 est en général inactivée par la protéine E6 virale, alors que dans les cancers HPV-, son inactivation résulte d’une mutation. Certaines études ont montré une corrélation inverse entre la présence de HPV et l’expression du récepteur de l’EGF (epidermal growth factor).
Les HPV ne peuvent pas être propagés in vitro, aussi le diagnostic virologique est-il le plus souvent moléculaire. Les outils disponibles font appel à différentes techniques de biologie moléculaire. L’absence de standardisation des modalités du diagnostic virologique des infections à HPV constitue un frein à la comparaison des résultats obtenus dans différentes études. La technique de génotypage « Inno-Lipa », ciblant un court fragment de 65 paires de bases, et donc plus adaptée aux prélèvements en paraffine qui sont archivés, est à ce jour la plus utilisée [18]. L’hybridation in situ (HIS) permet la caractérisation de l’ADN HPV en relation avec la morphologie cellulaire et tissulaire des tumeurs (Figure 3).
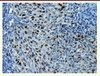 |
Figure 3. Hybridation in situ avec la sonde HPV. Présence d’un marquage dans les noyaux des cellules tumorales d’un carcinome épidermoïde (grossissement : x40). |
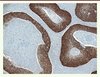 |
Figure 4. Étude immunohistochimique avec l’anticorps anti-p16. Il existe un marquage uniforme nucléaire et cytoplasmique de la majorité des cellules tumorales. Il s’agit d’un carcinome épidermoïde basaloïde typique (grossissement : x10). |
Une surexpression de p16 par la tumeur en immunohistochimie est un marqueur indirect d’une infection par HPV [19]. La protéine p16 est un inhibiteur des CDK4 et CDK6 (cyclin dependent kinases) qui inactivent pRb, et elle intervient dans la phase G1 du cycle cellulaire. pRb, codée par le gène RB1 situé sur le chromosome 13, fonctionne comme un régulateur négatif du cycle cellulaire en empêchant la progression des cellules en phase G1 et le passage du point de restriction. Elle se lie au facteur de transcription E2F et provoque son inhibition, empêchant ainsi l’expression de la cycline E et d’une série de gènes indispensables à la phase S. L’oncoprotéine virale E7, surexprimée par les virus oncogènes HPV, présente une forte affinité pour pRb et se lie principalement à sa forme hypophosphorylée (forme active). La dissociation du complexe Rb-E2F libère E2F sous sa forme active, ce qui permet le déblocage du cycle cellulaire et, par rétrocontrôle, une surexpression de p16 (Figures 2 et 4). Cette surexpression de p16, indépendante du génotype d’HPV, est commune aux tumeurs induites par HPV. Il existe une bonne corrélation entre ces trois marqueurs : la PCR, l’hybridation et la surexpression de p16.
HPV et cancers des VADS : données cliniques
Dans les CEVADS, la présence d’une infection par HPV semble être un facteur de bon pronostic. Cette évolution clinique favorable serait vraisemblablement due à une chimiosensiblité ou une radiosensibilité meilleures que celles des CEVADS non induits par les virus [19–21]. Cependant, les patients fumeurs HPV+ ont un moins bon pronostic que les patients HPV+ non fumeurs [22, 23]. Quelques travaux, plus rares, ont trouvé une corrélation inverse entre une infection par HPV et la survenue d’une récidive ou la survie [24, 25]. La surexpression de p16 par les cellules tumorales est aussi associée à un meilleur pronostic, même dans les tumeurs où une infection par HPV n’est pas retrouvée [26].
À ce jour, la prise en charge des tumeurs est la même quel que soit le statut HPV. La recherche d’une infection par HPV est cependant fortement recommandée dans les carcinomes oropharyngés, en particulier chez les patients jeunes, sans facteur de risque, afin de trouver un agent causal à leur pathologie.
HPV et cancers des VADS : modifications du système immunitaire
Différentes études ont montré qu’une réponse immunitaire contre le virus HPV est observée chez les patients atteints de cancers des VADS associés à HPV [27]. Ainsi, des lymphocytes T CD8+ reconnaissant des peptides dérivés des protéines E2, E6 et E7 d’HPV16 ont été mis en évidence, aussi bien dans le sang que dans la tumeur de ces patients [28–30]. Une infiltration lymphocytaire importante, ainsi qu’un profil transcriptomique reflétant une réponse adaptative (lymphocytes T spécifiques d’antigènes), ont été également détectés [31, 32]. Il existe donc bien une réponse immunitaire contre le virus HPV chez ces patients. Néanmoins, cette réponse n’est pas optimale sur le plan fonctionnel, car ces lymphocytes T produisent peu d’IFNγ (interféron γ)et leur activité cytotoxique antitumorale est faible [30]. Les protéines E6 et E7 d’HPV inhibent la production d’IFNγ et d’IFNk par les cellules dendritiques [33, 34] et la voie de signalisation des Toll like receptor (TLR). Par ailleurs, il est connu que les cellules tumorales dans les CVADS produisent de nombreuses molécules immunosuppressives, mais aussi des facteurs pro-inflammatoires inhibant cette réponse antitumorale [27, 35].
Vaccination anti-HPV
Deux vaccins prophylactiques contre le papillomavirus, reposant sur l’immunogénicité de la protéine L1, ont été récemment commercialisés. Ils ciblent essentiellement les adolescentes en prévention du cancer et des lésions précancéreuses du col de l’utérus (une infection par un HPV de haut risque est retrouvée dans 90,7 % à 96,6 % des cancers du col) [43] ; le cancer du col de l’utérus représentant 10 % des cancers de la femme dans le monde et la deuxième cause de décès par cancer après le cancer du sein [43]. Le Gardasil (Sanofi-Pasteur - MSD) est un vaccin tétravalent protégeant contre les génotypes 16, 18, 6 et 11, tandis que le vaccin Cervarix (GlaxoSmithKline) bivalent est efficace contre les génotypes 16 et 18 du virus HPV. Ces deux vaccins sont efficaces chez les personnes qui n’ont jamais été en contact avec HPV. Ils sont recommandés en France chez les adolescentes, tandis qu’aux États-Unis le CDC (Center disease control and prevention) à Atlanta a étendu son indication aux jeunes garçons. L’augmentation croissante des cancers de l’oropharynx liés à HPV et des cancers de l’anus est à la base de cette recommandation américaine. Comme le génotype HPV16 est très fréquent (plus de 85 %) dans les CEVADS, l’impact de ces vaccins pourrait être encore plus important que pour la prévention du cancer du col de l’utérus (les deux génotypes 16 et/ou 18 ne sont retrouvés que dans 70 % des cas). Néanmoins, il a été montré que ces vaccins pouvaient protéger contre des génotypes d’HPV non présents dans le vaccin (protection croisée) [36].
Étant donné que ces vaccins ne sont efficaces qu’avant exposition au virus HPV et qu’on estime à plus de 500 millions le nombre de personnes infectées par des HPV oncogènes, différents groupes ont essayé de développer des vaccins thérapeutiques chez des patients présentant des cancers HPV+. Des résultats encourageants ont été obtenus chez des patientes atteintes de dysplasie vulvaire avec un vaccin cherchant à induire une réponse lymphocytaire T, tandis que les vaccins prophylactiques reposent sur l’induction d’anticorps [37, 38]. Néanmoins, ces vaccins n’ont pas démontré leur efficacité sur les carcinomes infiltrants HPV+. Le groupe de Melief a montré qu’au cours de la progression des tumeurs HPV+, il existait une augmentation des lymphocytes T régulateurs qui pourrait expliquer l’inhibition de la réponse lymphocytaire T anti-HPV induite par le vaccin [39]. Différentes stratégies sont développées par notre groupe et d’autres, pour contrecarrer ces mécanismes de résistance en associant à ces vaccins thérapeutiques des molécules - ou autres approches - qui lèveraient cette immunosuppression induite par les cancers [40, 41]. Il est intéressant de mentionner que la radiothérapie et certaines chimiothérapies (cisplatine par exemple), qui sont les traitements de référence des CEVADS, améliorent l’efficacité vaccinale des vaccins thérapeutiques anti-HPV [42]. Une réflexion est engagée sur la possibilité d’une désescalade thérapeutique chez les patients atteints de cancers des VADS, et de nouvelles approches thérapeutiques en cours d’exploration évaluent l’intérêt de combiner ces vaccins aux traitements de référence.
Conclusion
La responsabilité des virus HPV dans la genèse et le pronostic des CEVADS, notamment dans leur localisation oropharyngée, est maintenant démontrée. De nouvelles questions se posent aujourd’hui quant à la prise en charge de ces cancers : faut-il rechercher une infection HPV de façon systématique ? Faut-il proposer une prise en charge particulière de ces cancers qui concernent des patients plus jeunes et qui sont plus radiosensibles ? La présence d’HPV est-elle vraiment associée à un meilleur pronostic dans toutes les localisations et quel que soit le sous-type histologique ? Ne faut-il pas rediscuter de façon plus générale l’indication de la vaccination des garçons ?
L’étude et la meilleure compréhension de la susceptibilité génétique des infections à HPV, des relations hôtes-virus et aussi de la réponse immune anti-HPV dans la physiopathologie du cancer des VADS seront, sans aucun doute, des questions essentielles à résoudre pour une meilleure prise en charge de ces tumeurs dans les années à venir.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Références
- Cohen EE. Novel therapeutic targets in squamous cell carcinoma of the head and neck. Semin Oncol 2004 ; 31 : 755–768. [CrossRef] [PubMed] [Google Scholar]
- Syrjanen K, Syrjanen S, Lamberg M, et al. Morphological and immunohistochemical evidence suggesting human papillomavirus (HPV) involvement in oral squamous cell carcinogenesis. Int J Oral Surg 1983 ; 12 : 418–424. [CrossRef] [PubMed] [Google Scholar]
- Ryerson AB, Peters ES, Coughlin SS, et al. Burden of potentially human papillomavirus-associated cancers of the oropharynx and oral cavity in the US, 1998–2003. Cancer 2008 ; 113 : 2901–2909. [CrossRef] [PubMed] [Google Scholar]
- Hansson BG, Rosenquist K, Antonsson A, et al. Strong association between infection with human papillomavirus and oral and oropharyngeal squamous cell carcinoma: a population-based case-control study in southern Sweden. Acta Otolaryngol 2005 ; 125 : 1337–1344. [CrossRef] [PubMed] [Google Scholar]
- Syrjanen S. The role of human papillomavirus infection in head and neck cancers. Ann Oncol 2010 ; 21 : vii243–vii245. [CrossRef] [PubMed] [Google Scholar]
- Chaturvedi AK, Engels EA, Anderson WF, Gillison ML. Incidence trends for human papillomavirus-related and -unrelated oral squamous cell carcinomas in the United States. J Clin Oncol 2008 ; 26 : 612–619. [CrossRef] [PubMed] [Google Scholar]
- Kreimer AR, Clifford GM, Boyle P, Franceschi S. Human papillomavirus types in head and neck squamous cell carcinomas worldwide: a systematic review. Cancer Epidemiol Biomarkers Prev 2005 ; 14 : 467–475. [CrossRef] [PubMed] [Google Scholar]
- Dayyani F, Etzel CJ, Liu M, et al. Meta-analysis of the impact of human papillomavirus (HPV) on cancer risk, overall survival in head, neck squamous cell carcinomas (HNSCC). Head Neck Oncol 2010 ; 2 : 15. [Google Scholar]
- St Guily JL, Jacquard AC, Pretet JL, et al. Human papillomavirus genotype distribution in oropharynx and oral cavity cancer in France - The EDiTH VI study. J Clin Virol 2011 ; 51 : 100–104. [CrossRef] [PubMed] [Google Scholar]
- Hocking JS, Stein A, Conway EL, et al. Head and neck cancer in Australia between 1982 and 2005 show increasing incidence of potentially HPV-associated oropharyngeal cancers. Br J Cancer 2011 ; 104 : 886–891. [CrossRef] [PubMed] [Google Scholar]
- Si-Mohamed A, Badoual C, Hans S, et al. An unusual human papillomavirus type 82 detection in laryngeal squamous cell carcinoma: case report and review of literature. J Clin Virol 2012 ; 54 : 190–193. [CrossRef] [PubMed] [Google Scholar]
- D’Souza G, Kreimer AR, Viscidi R, et al. Case-control study of human papillomavirus and oropharyngeal cancer. N Engl J Med 2007 ; 356 : 1944–1956. [CrossRef] [PubMed] [Google Scholar]
- Gillison ML, Broutian T, Pickard RK, et al. Prevalence of oral HPV infection in the United States, 2009–2010. JAMA 2012 ; 307 : 693–703. [CrossRef] [PubMed] [Google Scholar]
- Chen R, Sehr P, Waterboer T, et al. Presence of DNA of human papillomavirus 16 but no other types in tumor-free tonsillar tissue. J Clin Microbiol 2005 ; 43 : 1408–1410. [CrossRef] [PubMed] [Google Scholar]
- Duray A, Descamps G, Bettonville M, et al. High prevalence of high-risk human papillomavirus in palatine tonsils from healthy children and adults. Otolaryngol Head Neck Surg 2011 ; 145 : 230–235. [CrossRef] [PubMed] [Google Scholar]
- Chernock RD, Lewis JS, Jr, Zhang Q, El-Mofty SK. Human papillomavirus-positive basaloid squamous cell carcinomas of the upper aerodigestive tract: a distinct clinicopathologic and molecular subtype of basaloid squamous cell carcinoma. Hum Pathol 2010 ; 41 : 1016–1023. [CrossRef] [PubMed] [Google Scholar]
- Thariat J, Badoual C, Faure C, et al. Basaloid squamous cell carcinoma of the head and neck: role of HPV and implication in treatment and prognosis. J Clin Pathol 2010 ; 63 : 857–866. [CrossRef] [PubMed] [Google Scholar]
- Shi W, Kato H, Perez-Ordonez B, et al. Comparative prognostic value of HPV16 E6 mRNA compared with in situ hybridization for human oropharyngeal squamous carcinoma. J Clin Oncol 2009 ; 27 : 6213–6221. [CrossRef] [PubMed] [Google Scholar]
- Shoushtari AN, Rahimi NP, Schlesinger DJ, Read PW. Survey on human papillomavirus/p16 screening use in oropharyngeal carcinoma patients in the United States. Cancer 2010 ; 116 : 514–519. [CrossRef] [PubMed] [Google Scholar]
- Fakhry C, Westra WH, Li S, et al. Improved survival of patients with human papillomavirus-positive head and neck squamous cell carcinoma in a prospective clinical trial. J Natl Cancer Inst 2008 ; 100 : 261–269. [CrossRef] [PubMed] [Google Scholar]
- Worden FP, Kumar B, Lee JS, et al. Chemoselection as a strategy for organ preservation in advanced oropharynx cancer: response and survival positively associated with HPV16 copy number. J Clin Oncol 2008 ; 26 : 3138–3146. [CrossRef] [PubMed] [Google Scholar]
- Ang KK, Harris J, Wheeler R, et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N Engl J Med 2010 ; 363 : 24–35. [Google Scholar]
- Maxwell JH, Kumar B, Feng FY, et al. Tobacco use in human papillomavirus-positive advanced oropharynx cancer patients related to increased risk of distant metastases and tumor recurrence. Clin Cancer Res 2010 ; 16 : 1226–1235. [CrossRef] [PubMed] [Google Scholar]
- Duray A, Descamps G, Decaestecker C, et al. Human papillomavirus DNA strongly correlates with a poorer prognosis in oral cavity carcinoma. Laryngoscope 2012 ; 122 : 1558–1565. [CrossRef] [PubMed] [Google Scholar]
- Rosenquist K, Wennerberg J, Annertz K, et al. Recurrence in patients with oral and oropharyngeal squamous cell carcinoma: human papillomavirus and other risk factors. Acta Otolaryngol 2007 ; 127 : 980–987. [CrossRef] [PubMed] [Google Scholar]
- Klingenberg B, Hafkamp HC, Haesevoets A, et al. p16 INK4A overexpression is frequently detected in tumour-free tonsil tissue without association with HPV. Histopathology 2010 ; 56 : 957–967. [CrossRef] [PubMed] [Google Scholar]
- Badoual C, Sandoval F, Pere H, et al. Better understanding tumor-host interaction in head and neck cancer to improve the design and development of immunotherapeutic strategies. Head Neck 2010 ; 32 : 946–958. [PubMed] [Google Scholar]
- Albers A, Abe K, Hunt J, et al. Antitumor activity of human papillomavirus type 16 E7-specific T cells against virally infected squamous cell carcinoma of the head and neck. Cancer Res 2005 ; 65 : 11146–11155. [CrossRef] [PubMed] [Google Scholar]
- Hoffmann TK, Arsov C, Schirlau K, et al. T cells specific for HPV16 E7 epitopes in patients with squamous cell carcinoma of the oropharynx. Int J Cancer 2006 ; 118 : 1984–1991. [CrossRef] [PubMed] [Google Scholar]
- Heusinkveld M, Welters MJ, van Poelgeest MI, et al. The detection of circulating human papillomavirus-specific T cells is associated with improved survival of patients with deeply infiltrating tumors. Int J Cancer 2011 ; 128 : 379–389. [CrossRef] [PubMed] [Google Scholar]
- Thurlow JK, Pena Murillo CL, Hunter KD, et al. Spectral clustering of microarray data elucidates the roles of microenvironment remodeling and immune responses in survival of head and neck squamous cell carcinoma. J Clin Oncol 2010 ; 28 : 2881–2888. [CrossRef] [PubMed] [Google Scholar]
- Kong CS, Narasimhan B, Cao H, et al. The relationship between human papillomavirus status and other molecular prognostic markers in head and neck squamous cell carcinomas. Int J Radiat Oncol Biol Phys 2009 ; 74 : 553–561. [CrossRef] [PubMed] [Google Scholar]
- Rincon-Orozco B, Halec G, Rosenberger S, et al. Epigenetic silencing of interferon-kappa in human papillomavirus type 16-positive cells. Cancer Res 2009 ; 69 : 8718–8725. [CrossRef] [PubMed] [Google Scholar]
- Koromilas AE, Li S, Matlashewski G. Control of interferon signaling in human papillomavirus infection. Cytokine Growth Factor Rev 2001 ; 12 : 157–170. [CrossRef] [PubMed] [Google Scholar]
- Badoual C, Bouchaud G, Agueznay Nel H, et al. The soluble alpha chain of interleukin-15 receptor: a proinflammatory molecule associated with tumor progression in head and neck cancer. Cancer Res 2008 ; 68 : 3907–3914. [CrossRef] [PubMed] [Google Scholar]
- Lehtinen M, Paavonen J, Wheeler CM, et al. Overall efficacy of HPV-16/18 AS04-adjuvanted vaccine against grade 3 or greater cervical intraepithelial neoplasia: 4-year end-of-study analysis of the randomised, double-blind PATRICIA trial. Lancet Oncol 2012 ; 13 : 89–99. [CrossRef] [PubMed] [Google Scholar]
- Kenter GG, Welters MJ, Valentijn AR, et al. Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N Engl J Med 2009 ; 361 : 1838–1847. [CrossRef] [PubMed] [Google Scholar]
- Daayana S, Elkord E, Winters U, et al. Phase II trial of imiquimod and HPV therapeutic vaccination in patients with vulval intraepithelial neoplasia. Br J Cancer 2010 ; 102 : 1129–1136. [CrossRef] [PubMed] [Google Scholar]
- Welters MJ, Kenter GG, van Steenwijk PJ, et al. Success or failure of vaccination for HPV16-positive vulvar lesions correlates with kinetics and phenotype of induced T-cell responses. Proc Natl Acad Sci USA 2010 ; 107 : 11895–11899. [CrossRef] [Google Scholar]
- Berraondo P, Nouze C, Preville X, et al. Eradication of large tumors in mice by a tritherapy targeting the innate, adaptive, and regulatory components of the immune system. Cancer Res 2007 ; 67 : 8847–8855. [CrossRef] [PubMed] [Google Scholar]
- Pere H, Montier Y, Bayry J, et al. A CCR4 antagonist combined with vaccines induces antigen-specific CD8+ T cells and tumor immunity against self antigens. Blood 2011 ; 118 : 4853–4862. [CrossRef] [PubMed] [Google Scholar]
- Van der Burg SH, Melief CJ. Therapeutic vaccination against human papilloma virus induced malignancies. Curr Opin Immunol 2011 ; 23 : 252–257. [CrossRef] [PubMed] [Google Scholar]
- Silbermann B, Launay O. Prévention des infections à papillomavirus et du zona : nouveaux vaccins. Med Sci (Paris) 2007 ; 23 : 423–427. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
Liste des figures
|
Figure 1. Représentation du génome HPV-16. Les phases ouvertes de lecture sont nommées E1, E2, E4, E5, E6 et E7 pour les protéines précoces (E pour early) et L1 et L2 pour les protéines tardives (L pour late). La région LCR (locus control region) contient les éléments de régulation de la transcription et de la régulation. |
| Dans le texte | |
|
Figure 2. Interférence de la protéine virale E7 avec le point de contrôle G1-S de la mitose. pRb se lie au facteur de transcription E2F et provoque son inhibition, empêchant ainsi l’expression de la cycline E et une série de gènes indispensables à la phase S. L’oncoprotéine virale E7, produite par les virus oncogènes HPV, présente une forte affinité pour pRb et se lie particulièrement à la forme hypophosphorylée (la forme active). La dissociation du complexe pRB-E2F libère E2F sous sa forme active, ce qui permet le déblocage du cycle cellulaire et entraîne la surexpression de p16, qui est une protéine de rétrocontrôle. |
| Dans le texte | |
|
Figure 3. Hybridation in situ avec la sonde HPV. Présence d’un marquage dans les noyaux des cellules tumorales d’un carcinome épidermoïde (grossissement : x40). |
| Dans le texte | |
|
Figure 4. Étude immunohistochimique avec l’anticorps anti-p16. Il existe un marquage uniforme nucléaire et cytoplasmique de la majorité des cellules tumorales. Il s’agit d’un carcinome épidermoïde basaloïde typique (grossissement : x10). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.