")
")
| Issue |
Med Sci (Paris)
Volume 28, Number 12, Décembre 2012
|
|
|---|---|---|
| Page(s) | 1032 - 1034 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/20122812003 | |
| Published online | 21 December 2012 | |
Contrôle épigénétique de la stabilité phénotypique et fonctionnelle des lymphocytes Th2 par la voie Suv39h1/HP1α
Epigenetic control of Th2 helper cell differentiation by the Suv39h1/HP1α pathway
1
Institut Curie, 26, rue d’Ulm, 75005 Paris, France
2
Inserm U932, Paris, France
3
CNRS UMR218, Paris, France
4
Institut de recherche en cancérologie de Montpellier, Centre régional de lutte contre le cancer (CRLC) Val d’Aurelle-Paul Lamarque, 34298 Montpellier Cedex 5, France
5
Adresse actuelle : division of molecular immunology, Walter and Eliza Hall institute of medical research, Melbourne, Australie
6
Adresse actuelle : laboratory of molecular immunology and Rockefeller university, New York, États-Unis
7
Adresse actuelle : laboratoire de protéomique et de spectrométrie de masse, Institut Curie, Paris, 26, rue d’Ulm 75005 Paris, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Ces auteurs ont contribué de manière égale à cet article.
Les lymphocytes T naïfs (qui n’ont pas encore été activés) peuvent se différencier en sous-populations de cellules effectrices selon le type de pathogène qu’ils rencontrent et l’environnement dans lequel ils se trouvent [1]. Les principaux sous-types de lymphocytes T CD4+ sont appelés Th1, Th2, Th17 ou T régulateurs (Treg). Ils sont caractérisés par des profils de sécrétion de cytokines distincts, adaptés à leurs fonctions respectives. Les lymphocytes Th2 par exemple produisent de l’interleukine 4 (IL-4), de l’IL-5 et de l’IL-13, nécessaires à la commutation de classe des lymphocytes B (qui correspond au changement de classe - μ1 [IgM] vers γ1 [IgG] par exemple - de la chaîne lourde des immunoglobulines au cours de réarrangements par recombinaison génétique), alors que les cellules Th1 sont caractérisées par la sécrétion d’interféron γ (IFNγ) qui est impliqué, entre autres, dans l’activation de l’activité bactéricide des phagocytes et dans la différenciation des lymphocytes T CD8+ en cellules T cytotoxiques. La différenciation des lymphocytes est induite par des combinaisons de cytokines spécifiques à chaque lignage (IL-12 pour les Th1, IL-4 pour les Th2, IL-6/TGF-β [transforming growth factor β] pour les Th17 et IL-2/ TGF-β pour les Treg). Ces médiateurs solubles induisent l’expression de facteurs de transcription (T-bet pour les Th1, GATA-3 [transactivating T-cell-specific transcription factor] pour les Th2, RORγt [RAR-related orphan receptor gamma] pour les Th17 et FoxP3 [forkhead box P3] pour les Treg) qui orchestreront la mise en place de programmes d’expression génique spécifiques, et donc l’acquisition des fonctions effectrices [1].
Origine épigénétique de la perte de plasticité des lymphocytes T
Au cours du processus de différenciation, les lymphocytes T naïfs perdent progressivement leur plasticité (leur capacité à se transdifférencier en d’autres sous-types) [2] à des degrés différents selon les sous-types considérés. Les lymphocytes Th2, par exemple, sont très peu plastiques. Même dans un environnement pro-Th1 (en présence d’IL-12 par exemple), ils maintiennent leur phénotype et leurs fonctions, et ne se transdifférencient pas en lymphocytes Th1. À l’opposé, les lymphocytes Th17 conservent une certaine plasticité. Ces cellules ont notamment la capacité de se transdifférencier en cellules Th1 dans certaines situations physiopathologiques [1].
Les bases moléculaires de la perte de plasticité des lymphocytes T naïfs au cours de la différenciation sont encore mal comprises. L’intervention de facteurs épigénétiques a été proposée. Cette hypothèse s’appuie sur des études montrant que le phénotype acquis par un lymphocyte T activé est transmis aux cellules filles lors de la division cellulaire, même en absence des signaux environnementaux qui ont induit la différenciation. Plus récemment, l’observation de marques d’histones qualifiées de permissives, car associées aux gènes actifs [3, 4] et retrouvées au niveau des gènes spécifiques d’un lignage donné (codant pour les cytokines ou les facteurs de transcription spécifiques de ce lignage), a permis de proposer un mécanisme épigénétique à l’origine de cette perte de plasticité. D’autant plus que, de façon concomitante dans un même lignage, les gènes impliqués dans la différenciation des autres sous-types acquièrent des marques répressives (caractéristiques de gènes silencieux), suggérant qu’ils sont activement réprimés [3, 4]. Néanmoins, il reste à comprendre si ces changements de marquage épigénétique sont une cause ou une simple conséquence de l’état d’expression de ces gènes.
Le voie de répression épigénétique Suv39h1/HP1α
Nous nous sommes donc intéressés plus directement aux mécanismes d’activation et de suppression de l’expression de ces gènes au cours de la différenciation lymphocytaire, pour établir un lien entre les marques et le statut fonctionnel des gènes. Pour cela, nous avons exploré l’importance d’une voie de régulation impliquée dans l’organisation de l’hétérochromatine péricentrique et dans la répression transcriptionnelle. Cette voie implique l’histone méthyltransférase Suv39h et son partenaire HP1 (hétérochromatin protéin 1). HP1 a initialement été décrite chez la drosophile comme une protéine non-histone associée à l’hétérochromatine et impliquée dans la suppression de l’expression de gènes par effet de position. Le gène codant pour cette protéine est trouvé chez de nombreux organismes eucaryotes. La protéine elle-même est conservée d’un point de vue phylogénétique, tant au niveau de sa séquence primaire en acides aminés que de son organisation fonctionnelle. Chez les mammifères, trois isoformes ont été décrites : HP1α, β et γ [5]. HP1 se lie préférentiellement à la lysine 9 triméthylée de l’histone 3 (H3K9me3). Cette modification est mise en place par l’action coordonnée de différentes histones méthyltransférases, la principale étant l’enzyme Suv39h1. Dans l’hétérochromatine, la triméthylation de H3K9 est en effet imposée par Suv39h1, qui s’associe aussi à HP1α et ainsi favorise son ancrage sur les histones triméthylées. Suv39h1 intervient donc dans une boucle de régulation qui assure le maintien et la propagation de l’hétérochromatine [6–8]. Si la triméthylation de H3K9 constitue le point d’ancrage de HP1a sur la chromatine, il est important de noter que l’acétylation de ce résidu de l’histone 3 (H3K9ac), considérée comme une marque de transcription active, est à l’inverse défavorable à la liaison de HP1 [9].
Rôle de la voie Suv39h1/HP1α dans la stabilité phénotypique et fonctionnelle des lymphocytes Th2
Notre analyse de la méthylation et de l’acétylation de H3K9 au cours de la différenciation des lymphocytes T CD4+ naïfs en lymphocytes Th1 ou Th2 s’est avérée très informative. En effet, nous montrons que les gènes actifs dans les lymphocytes Th1 (codant pour l’IFNγ et T-bet) et Th2 (codant pour l’IL-4 et GATA-3) sont enrichis en H3K9 acétylée, alors que les gènes inactifs (codant pour l’IFNγ et T-bet dans les Th2, et pour l’IL-4 et GATA3 dans les Th1) sont majoritairement triméthylés sur H3K9. Afin de déterminer l’importance fonctionnelle de ces marques, nous avons répété ces expériences sur les lymphocytes chez des souris déficientes pour Suv39h1 (knock-out) [10]. Dans les cellules Th2 dépourvues de Suv39h1, nous avons alors observé que la proportion de H3K9me3 dans le promoteur et certaines régions régulatrices du gène codant pour l’IFNγ était diminuée, suggérant ainsi que Suv39h1 a un rôle clé dans l’établissement de cette marque. Quant aux niveaux d’acétylation de H3K9, ils augmentaient sur ces mêmes positions et aussi sur le promoteur du gène codant pour T-bet. Pour établir que les différences de marques épigénétiques entre cellules contrôles et Suv39h1-KO ne se traduisent pas par des différences phénotypiques ou fonctionnelles, nous avons réalisé des expériences de différenciation in vitro de lymphocytes T naïfs en cellules Th1 ou Th2. Nos résultats indiquent que les modifications de H3K9 n’ont pas de rôle majeur dans le contrôle de l’expression de ces gènes au cours de la différenciation primaire des cellules T naïves en lymphocytes Th1 ou Th2.
À ce stade, il était intéressant de tester si les différences observées pouvaient avoir un impact sur la plasticité des lymphocytes. Nous avons donc cultivé dans un environnement Th1 des cellules Th2 différenciées in vitro. Contrairement aux cellules contrôles qui sont incapables d’exprimer T-bet ou de produire de l’IFNγ, nous avons observé qu’une proportion importante de cellules déficientes pour Suv39h1 sont capables d’exprimer ces gènes. Ces résultats montrent qu’en absence de Suv39h1, les lymphocytes Th2 peuvent se différencier vers le phénotype Th1, plasticité qui est perdue dans les cellules sauvages. Nous avons émis l’hypothèse que ce gain de plasticité dans les cellules Th2 différenciées Suv39h1-KO pourrait être dû au déséquilibre de la proportion entre la triméthylation et l’acétylation dans les régions régulatrices des gènes codant pour l’IFNγ et T-bet. Ainsi, les changements de marques épigénétiques observés, conséquence de l’absence de Suv39h1, seraient donc responsables de l’absence de suppression active de ces gènes dans les cellules Suv39h1-KO. Nous avons montré que HP1α, l’un des partenaires les plus importants dans la suppression génique induite par Suv39h1, est également présente sur les régions promotrices des gènes codant pour l’IFNγ et T-bet dans les cellules Th2, et son recrutement nécessite l’expression de Suv39h1.
Afin de tester l’importance fonctionnelle de HP1α, nous avons généré des souris déficientes pour cette protéine. Comme dans le cas des lymphocytes T déficients pour Suv39h1, les lymphocytes T naïfs CD4+ déficients pour HP1a se différencient normalement en effecteurs Th1 ou Th2, et les lymphocytes Th2 présentent une plasticité accrue lorsqu’ils sont placés dans un environnement Th1. Ces résultats sont en accord avec notre hypothèse selon laquelle dans les lymphocytes Th2, la suppression active des gènes caractéristiques du lignage Th1 est induite par la voie H3K9me3/Suv39h1/HP1a. Lorsque cette voie n’est pas active, la répression de ces gènes est incomplète et ils peuvent être réexprimés si les cellules sont placées dans un environnement adéquat (Figure 1).
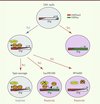 |
Figure 1. Modèle hypothétique de régulation de la suppression de l’expression du gène de l’IFN γ dans les lymphocytes Th2. Lors de la différenciation des lymphocytes T naïfs en Th1, les régions régulatrices du gène codant pour l’IFNγ (qui est exprimé activement) sont enrichies en H3K9ac. Dans les lymphocytes Th2, en revanche, le gène codant pour l’IFNγ est inactif et certaines régions régulatrices sont enrichies en H3K9me3. Lorsque les lymphocytes Th2 sont mis dans un environnement Th1, le gène de l’IFNγ n’est pas réexprimé car il est inactivé par le recrutement de HP1a. En l’absence de Suv39h1, le niveau de H3K9me3 dans le gène de l’IFNγ diminue alors que le niveau de H3K9ac augmente. HP1α ne peut plus être recrutée efficacement, ce qui provoque un défaut dans l’inactivation et la réexpression du gène codant pour l’IFNγ. De même, en l’absence de HP1α, l’inactivation du gène de l’IFNγ dans les lymphocytes Th2 est incomplète, et le gène est réexprimé si les lymphocytes Th2 sont mis dans un environnement Th1. |
Conséquences en pathologie de la répression épigénétique dans les Th2
Afin d’évaluer la signification physiopathologique de cette voie de régulation épigénétique, nous nous sommes penchés sur un modèle d’asthme allergique chez la souris. Ce modèle est caractérisé par l’induction d’une forte réponse Th2 chez les souris témoins, à l’origine d’une réponse inflammatoire dérégulée aboutissant à l’obstruction des voies respiratoires. Dans ce modèle, nos expériences ont permis de montrer que, contrairement à ce qui est observé chez les souris témoins, des lymphocytes Th1 et mixtes Th1/Th2 (produisant à la fois de l’IFNγ et de l’IL-4) se différencient chez les souris déficientes pour Suv39h1. Cette réversion de la réponse immune vers un phénotype Th1 se traduit par une diminution de l’inflammation dans les poumons. Ces résultats suggèrent qu’en absence de Suv39h1, la différenciation des lymphocytes T naïfs en cellules effectrices Th2 est moins stricte. Les lymphocytes Th1, qui peuvent alors se différencier, ont un rôle protecteur [11].
Nos résultats montrent donc que la voie de répression épigénétique Suv39h1/HP1α est nécessaire à la stabilité phénotypique et fonctionnelle des lymphocytes Th2 in vitro et in vivo. Ces résultats ouvrent des perspectives thérapeutiques intéressantes en s’appuyant sur des inhibiteurs pharmacologiques de Suv39h1. En effet, nous avons montré que de telles molécules réduisent la sévérité de la pathologie dans le modèle d’asthme allergique que nous avons utilisé.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Références
- Wilson CB, Rowell E, Sekimata M. Epigenetic control of T-helper-cell differentiation. Nat Rev Immunol 2009 ; 9 : 91–105. [CrossRef] [PubMed] [Google Scholar]
- Grogan JL, Mohrs M, Harmon B, et al. Early transcription and silencing of cytokine genes underlie polarization of T helper cell subsets. Immunity 2001 ; 14 : 205–215. [CrossRef] [Google Scholar]
- Avni O, Macian F, Szabo SJ, et al. T(H) cell differentiation is accompanied by dynamic changes in histone acetylation of cytokine genes. Nat Immunol 2002 ; 3 : 643–651. [CrossRef] [PubMed] [Google Scholar]
- Wei G, Wei L, Zhu J, et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity 2009 ; 30 : 155–167. [CrossRef] [PubMed] [Google Scholar]
- Kwon SH, Workman JL. The heterochromatin protein 1 (HP1) family: put away a bias toward HP1. Mol Cell 2008 ; 26 : 217–224. [Google Scholar]
- Maison C, Almouzni G. HP1 and the dynamics of heterochromatin maintenance. Nat Rev Mol Cell Biol 2004 ; 5 : 296–304. [CrossRef] [PubMed] [Google Scholar]
- Lachner M, O’Carroll D, Rea S, et al. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 2001 ; 410 : 116–120. [CrossRef] [PubMed] [Google Scholar]
- Bannister AJ, Zegerman P, Partridge JF, et al. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 2001 ; 410 : 120–124. [CrossRef] [PubMed] [Google Scholar]
- Mateescu B, England P, Halgand F, et al. Tethering of HP1 proteins to chromatin is relieved by phosphoacetylation of histone H3. EMBO Rep 2004 ; 5 : 490–496. [CrossRef] [PubMed] [Google Scholar]
- Peters AH, O’Carroll D, Scherthan H, et al. Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell 2001 ; 107 : 323–337. [CrossRef] [PubMed] [Google Scholar]
- Barnes PJ. Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol 2008 ; 8 : 183–192. [CrossRef] [PubMed] [Google Scholar]
© 2012 médecine/sciences – Inserm / SRMS
Liste des figures
|
Figure 1. Modèle hypothétique de régulation de la suppression de l’expression du gène de l’IFN γ dans les lymphocytes Th2. Lors de la différenciation des lymphocytes T naïfs en Th1, les régions régulatrices du gène codant pour l’IFNγ (qui est exprimé activement) sont enrichies en H3K9ac. Dans les lymphocytes Th2, en revanche, le gène codant pour l’IFNγ est inactif et certaines régions régulatrices sont enrichies en H3K9me3. Lorsque les lymphocytes Th2 sont mis dans un environnement Th1, le gène de l’IFNγ n’est pas réexprimé car il est inactivé par le recrutement de HP1a. En l’absence de Suv39h1, le niveau de H3K9me3 dans le gène de l’IFNγ diminue alors que le niveau de H3K9ac augmente. HP1α ne peut plus être recrutée efficacement, ce qui provoque un défaut dans l’inactivation et la réexpression du gène codant pour l’IFNγ. De même, en l’absence de HP1α, l’inactivation du gène de l’IFNγ dans les lymphocytes Th2 est incomplète, et le gène est réexprimé si les lymphocytes Th2 sont mis dans un environnement Th1. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.