")
")
| Issue |
Med Sci (Paris)
Volume 28, Number 6-7, Juin–Juillet 2012
|
|
|---|---|---|
| Page(s) | 565 - 568 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/2012286002 | |
| Published online | 16 juillet 2012 | |
Transmission interespèces des prions
Le tissu lymphoïde s’en mêle
Tissue-specific cross-species transmission of prions
1
UR892 Inra (Institut national de la recherche agronomique), virologie et immunologie moléculaires, 78350 Jouy-en Josas, France
2
UMR1313 INRA, génétique animale et biologie intégrative, 78350 Jouy-en-Josas, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.

Photo : prions (en vert) visualisés par immunofluorescence dans une rate de souris (en rouge les lymphocytes B de la rate) (© INRA/VIM/V. Béringue).
Un agent infectieux de nature protéique
Le prion est le pathogène responsable, chez les mammifères, de désordres neurodégénératifs d’issue fatale dénommés encéphalopathies spongiformes transmissibles (EST) ou maladies à prion. Les EST affectent à la fois l’homme (maladie de Creutzfeldt-Jakob [MCJ]) et les animaux de rente ou sauvages (tremblante du mouton, encéphalopathie spongiforme bovine [ESB], maladie du dépérissement chronique des cervidés [MDC]).
Les prions sont essentiellement composés d’agrégats d’une forme anormalement repliée (PrPSc) d’une protéine de l’hôte, la protéine prion cellulaire (PrPC). La PrPC est une protéine membranaire glycosylée, exprimée de façon quasi ubiquiste dans l’organisme. Sa séquence en acides aminés est très conservée entre les espèces. Bien que son rôle physiologique ne soit pas formellement établi, sa présence apparaît importante au cours du développement embryonnaire [1, 2]. L’invalidation expérimentale de PrPC rend les animaux résistants aux prions. Lors des étapes initiales de l’infection, l’interaction physique entre les oligomères de PrPSc du prion infectant et la PrPC de l’hôte induirait son changement de conformation, notamment un enrichissement en feuillets β, enclenchant ainsi un cycle autoentretenu de multiplication des prions au cours duquel la PrPC produite par les cellules serait convertie par les molécules de PrPSc précédemment néosynthétisées [3]. Dans le cerveau, cela conduit à l’accumulation de PrPSc sous forme de dépôts de type amyloïde, contribuant ainsi aux désordres neurodégénératifs caractéristiques des EST. Bien que ce mode de propagation s’apparente à celui décrit dans d’autres protéinopathies telles que la maladie d’Alzheimer ou de Parkinson, il produit dans le cas des EST un agent authentiquement infectieux pour la même espèce ou d’autres espèces de mammifères.
Compatibilité conformationnelle et barrière d’espèce
Comme les pathogènes conventionnels, les prions arborent une diversité de souches au sein d’une même espèce-hôte. Ces souches se différencient par leurs propriétés biologiques (temps d’incubation chez l’hôte infecté, neuropathologie) et physicochimiques (profil électrophorétique, résistance de la PrPSc à des agents dénaturants). En l’absence d’acides nucléiques spécifiques, la diversité des souches tiendrait à la capacité de la PrPSc d’exister sous plusieurs conformations stables, capables de transmettre fidèlement une information biologique spécifique [4]. Le support structural de cette information biologique reste à déterminer. Il pourrait impliquer la structure tertiaire et/ou quaternaire de la protéine [5].
Les prions peuvent également se transmettre d’une espèce à une autre et présenter un risque zoonotique : le variant de la MCJ apparu en 1996 chez l’homme lors de la crise dite de la « vache folle » provient de l’ingestion d’aliments contaminés par les prions ESB. La capacité des prions à se propager entre espèces différentes est limitée par une barrière, communément appelée barrière d’espèce. La recherche de signes neurologiques typiques de la présence des EST et de PrPSc dans le cerveau est le moyen classiquement utilisé pour apprécier la porosité de cette barrière, l’absence concomitante de ces marqueurs traduisant la faible réceptivité apparente des animaux inoculés. Au plan moléculaire, la transmission interespèces reposerait sur le degré de compatibilité spatiale entre les conformations de la PrPC de l’hôte et de la PrPSc du prion infectant (la souche). Intrinsèquement, la PrPC de l’hôte pourrait n’adopter qu’un nombre fini de conformations dans sa forme PrPSc pathologique. La barrière d’espèce sera faible si la PrPSc infectante appartient à ce panel de conformations possibles. Sinon, la barrière sera forte et ne sera éventuellement franchie que via l’émergence d’un « mutant » compatible susceptible de présenter de nouvelles propriétés biologiques [4, 6]. À l’inverse, l’expression chez une souris transgénique du gène Prnp (codant pour PrPC) de l’espèce hétérologue donneuse de prion permet d’estomper la barrière de transmission entre cette espèce et la souris (Figure 1).
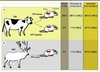 |
Figure 1. Efficacité de la transmission interespèces des prions en fonction du tissu. Les souris transgéniques exprimant la PrPC bovine sont parfaitement susceptibles à l’agent responsable de l’encéphalopathie spongiforme bovine (ESB) (A) . En revanche, les souris transgéniques exprimant la PrPC humaine ne développent pas de symptômes cliniques après inoculation du même agent (A) . Il en va de même pour les souris transgéniques exprimant la PrPC ovine inoculées par l’agent responsable de la maladie du dépérissement chronique des cervidés (MDC) (B) . Signe supplémentaire d’une forte barrière d’espèce apparente, un nombre très limité de souris transgéniques pour la PrPC ovine et humaine, toutes en fin de vie, accumulent de la protéine prion pathologique dans le cerveau (AB) . Néanmoins, une grande majorité de souris analysées présente des taux détectables de protéine prion pathologique dans la rate, et ce dès une année postinfection (AB) . |
Tissu extraneural et transmission interespèces
À partir de ce modèle de compatibilité conformationnelle s’est posée la question de l’influence du tissu d’un même hôte sur l’efficacité de transmission interspécifique des prions, du fait par exemple de variations moléculaires de la protéine PrPC ou d’un environnement cellulaire différent. Le tissu lymphoïde était déjà connu pour être un lieu de transit, quelle que soit la voie d’entrée du prion dans l’organisme [12], et être permissif à un certain nombre de souches lors de transmissions intraespèces. Nous nous sommes donc focalisés sur ce tissu et avons étudié sa permissivité relative par rapport au tissu nerveux lors de transmissions interespèces en apparence infructueuses, destinées à modéliser une transmission bovin-homme ou cervidé-ovin [7]. Lorsque les prions responsables de la MDC ou de l’ESB ont été injectés dans le cerveau de souris transgéniques pour la PrPC ovine et humaine et non plus murine, les prions se sont quasiment exclusivement multipliés dans le tissu lymphoïde, en l’occurrence la rate, et ce relativement précocement au regard de la durée de vie de l’animal (Figures 1 et 2). Quantitativement, la barrière d’espèce s’est avérée être environ dix fois moins étanche dans la rate que dans le cerveau (Figures 1 et 2). De plus, l’inoculation à la souris transgénique pour la PrPC ovine de prions de hamster (souche Sc237) a dévoilé que cette facilitation de la réplication splénique était indépendante du lymphotropisme intrinsèque de l’agent inoculé. L’adaptation sériée de cet agent Sc237 sur la séquence ovine a aussi permis de montrer que, lorsque la barrière d’espèce a finalement été franchie par l’émergence d’un prion « mutant » dans le cerveau des animaux inoculés, un agent différent se propageait dans la rate, se distinguant notamment par sa capacité unique à réinfecter l’hôte de départ (souris transgéniques pour la PrPC du hamster) (Figure 3).
 |
Figure 2. Accumulation de la protéine prion pathologique (PrPSc) dans le tissu splénique et cérébral de souris transgéniques exprimant la PrP humaine et inoculées par l’agent responsable de l’ESB. Cinétique d’accumulation de la PrPSc dans la rate (rouge) et le cerveau (bleu) de souris transgéniques pour la PrPC humaine inoculées par quatre isolats d’ESB (symboles). Les souris sans symptômes cliniques ont été euthanasiées au cours de l’infection (trois souris par point) ou en fin de vie (> 550 jours). La protéine est détectée par immunoblot. Les symboles vides signalent l’absence de PrPSc détectable dans le tissu étudié. |
 |
Figure 3. Sélection préférentielle des prions en fonction du tissu lors de transmissions interespèces. Le passage sérié de prions de hamster Sc237 aux souris transgéniques pour la PrPC ovine conduit à l’individualisation de deux agents prion distincts dans la rate et le cerveau, comme attesté par les différences de profil électrophorétique (A) ou de distribution cérébrale (B) de la protéine PrPSc. Seul l’agent qui s’est préférentiellement propagé dans la rate est capable de réinfecter des souris transgéniques pour la PrPC de hamster (animaux marrons). |
Pourquoi la rate apparaît-elle plus permissive que le cerveau à un prion étranger ? Par analogie avec le concept de quasi-espèce en virologie1, les prions pourraient héberger un continuum de conformations de PrPSc [4]. Certaines pourraient-elles être plus ou moins compatibles avec les différents tissus ? Des facteurs spécifiques des tissus de l’hôte pourraient aussi réguler la dynamique de réplication du prion. Ainsi, la PrPC, du fait de variations de conformations, ou d’autres créant un environnement favorable, permettraient par exemple une persistance accrue et propice à l’initiation de la conversion hétérospécifique. Quel que soit le mécanisme mis en jeu, notre étude révèle une capacité d’adaptation insoupçonnée de ce pathogène neurotrope lors d’une pression sélective, lui permettant au final d’étendre son spectre d’hôte.
Perspectives pour la santé publique
La barrière d’espèce prions ESB-homme apparaît ainsi moins robuste qu’initialement anticipé avec des modèles expérimentaux humanisés focalisés sur la contamination du système nerveux central [8, 9]. Cela implique que les tissus extraneuronaux devraient être étudiés de manière systématique lors de l’évaluation expérimentale du potentiel zoonotique des prions animaux.
Nos données apportent également un éclairage nouveau quant aux divergences observées entre le nombre de cas de variant MCJ cliniques au Royaume-Uni (environ 175 à ce jour) et la prévalence estimée (> 15 000) à partir de la présence de prions dans des tissus lymphoïdes (amygdale, appendice) prélevés lors d’actes chirurgicaux routiniers [10, 11]. L’exposition humaine aux prions ESB pourrait conduire à une colonisation du tissu lymphoïde nettement plus large que celle du cerveau. Afin d’assurer au mieux le suivi de cette épidémie, la recherche à long terme de porteurs asymptomatiques apparaît d’autant plus importante que notre étude suggère que les prions peuvent se propager silencieusement pendant plus du tiers de la durée de vie de l’individu (Figure 2) avant d’être détectable en périphérie par les méthodes conventionnelles de diagnostic. D’où la nécessité de développer un test sanguin capable de révéler une infection silencieuse et d’éviter des transmissions secondaires par le sang ou les actes médicaux.
Liens d’Intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Remerciements
Le travail résumé dans cet article a bénéficié en partie du soutien d’INRA-transfert et de la Fondation Alliance Biosecure. Merci à Claire Laude pour l’iconographie.
Les virus à ARN expriment un taux élevé de mutations – dues aux erreurs de réplication - ce qui entraîne une grande hétérogénéité dans les populations, mais ces variants continuent d’évoluer comme s’ils ne formaient « quasiment qu’une seule et même espèce » (espèce au sens d’espèce chimique ou moléculaire, et non au sens d’espèce biologique).
Références
- Khalife M, Young R, Passet B, et al. Transcriptomic analysis brings new insight into the biological role of the prion protein during mouse embryogenesis. PLoS One 2011 ; 6 : e23253. [CrossRef] [PubMed] [Google Scholar]
- Malaga-Trillo E, Solis GP, Schrock Y, et al. Regulation of embryonic cell adhesion by the prion protein. PLoS Biol 2009 ; 7 : e55. [CrossRef] [PubMed] [Google Scholar]
- Caughey B, Baron GS, Chesebro B, Jeffrey M. Getting a grip on prions: oligomers, amyloids, and pathological membrane interactions. Annu Rev Biochem 2009 ; 78 : 177–204. [CrossRef] [PubMed] [Google Scholar]
- Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science 2007 ; 318 : 930–936. [CrossRef] [PubMed] [Google Scholar]
- Tixador P, Herzog L, Reine F, et al. The physical relationship between infectivity, prion protein aggregates is strain-dependent. PLoS Pathog 2010 ; 6 : e1000859. [CrossRef] [PubMed] [Google Scholar]
- Beringue V, Vilotte JL, Laude H. Prion agent diversity, species barrier. Vet Res 2008 ; 39 : 47. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Beringue V, Herzog L, Jaumain E, et al. Facilitated cross-species transmission of prions in extraneural tissue. Science 2012 ; 335 : 472–475. [CrossRef] [PubMed] [Google Scholar]
- Asante EA, Linehan JM, Desbruslais M, et al. BSE prions propagate as either variant CJD-like or sporadic CJD-like prion strains in transgenic mice expressing human prion protein. EMBO J 2002 ; 21 : 6358–6366. [CrossRef] [PubMed] [Google Scholar]
- Bishop MT, Hart P, Aitchison L, et al. Predicting susceptibility and incubation time of human-to-human transmission of vCJD. Lancet Neurol 2006 ; 5 : 393–398. [CrossRef] [PubMed] [Google Scholar]
- Health Protection Agency, UK. http://www.hpa.org.uk/hpr/archives/2011/news3611.htm. [Google Scholar]
- Hilton DA, Ghani AC, Conyers L, et al. Prevalence of lymphoreticular prion protein accumulation in UK tissue samples. J Pathol 2004 ; 203 : 733–739. [CrossRef] [PubMed] [Google Scholar]
- Bachy V, Aucouturier P. Maladies à prions. Quel rôle pour les cellules dendritiques dans la pathogenèse des formes transmises ? Med Sci (Paris) 2010 ; 26 : 615–620. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
© 2012 médecine/sciences – Inserm / SRMS
Liste des figures
|
Figure 1. Efficacité de la transmission interespèces des prions en fonction du tissu. Les souris transgéniques exprimant la PrPC bovine sont parfaitement susceptibles à l’agent responsable de l’encéphalopathie spongiforme bovine (ESB) (A) . En revanche, les souris transgéniques exprimant la PrPC humaine ne développent pas de symptômes cliniques après inoculation du même agent (A) . Il en va de même pour les souris transgéniques exprimant la PrPC ovine inoculées par l’agent responsable de la maladie du dépérissement chronique des cervidés (MDC) (B) . Signe supplémentaire d’une forte barrière d’espèce apparente, un nombre très limité de souris transgéniques pour la PrPC ovine et humaine, toutes en fin de vie, accumulent de la protéine prion pathologique dans le cerveau (AB) . Néanmoins, une grande majorité de souris analysées présente des taux détectables de protéine prion pathologique dans la rate, et ce dès une année postinfection (AB) . |
| Dans le texte | |
|
Figure 2. Accumulation de la protéine prion pathologique (PrPSc) dans le tissu splénique et cérébral de souris transgéniques exprimant la PrP humaine et inoculées par l’agent responsable de l’ESB. Cinétique d’accumulation de la PrPSc dans la rate (rouge) et le cerveau (bleu) de souris transgéniques pour la PrPC humaine inoculées par quatre isolats d’ESB (symboles). Les souris sans symptômes cliniques ont été euthanasiées au cours de l’infection (trois souris par point) ou en fin de vie (> 550 jours). La protéine est détectée par immunoblot. Les symboles vides signalent l’absence de PrPSc détectable dans le tissu étudié. |
| Dans le texte | |
|
Figure 3. Sélection préférentielle des prions en fonction du tissu lors de transmissions interespèces. Le passage sérié de prions de hamster Sc237 aux souris transgéniques pour la PrPC ovine conduit à l’individualisation de deux agents prion distincts dans la rate et le cerveau, comme attesté par les différences de profil électrophorétique (A) ou de distribution cérébrale (B) de la protéine PrPSc. Seul l’agent qui s’est préférentiellement propagé dans la rate est capable de réinfecter des souris transgéniques pour la PrPC de hamster (animaux marrons). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.