")
")
| Issue |
Med Sci (Paris)
Volume 28, Number 1, Janvier 2012
|
|
|---|---|---|
| Page(s) | 39 - 41 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/2012281014 | |
| Published online | 27 janvier 2012 | |
Rac1 est la cible de l’activité E3 ubiquitine-ligase du suppresseur de tumeur HACE1
The tumor suppressor HACE1 targets Rac1 to ubiquitin-mediated proteasomal degradation
1
Institut Cochin, département de génétique et développement, Inserm U1016, CNRS UMR8104, université Paris Descartes, 24, rue du Faubourg Saint Jacques, 75014 Paris, France
2
Inserm, U634, faculté de médecine de Nice, 27, avenue de Valombrose, 06107 Nice, France
3
Inserm, U895, centre méditerranéen de médecine moléculaire, C3M, toxines microbiennes dans la relation hôte-pathogènes, université de Nice-Sophia-Antipolis, UFR médecine, 06204 Nice, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
La petite GTPase Rac1 contrôle des processus cellulaires essentiels, au nombre desquels la migration, la phagocytose, le cycle et l’apoptose [1]. La dérégulation de l’activité de Rac1 est impliquée dans des pathologies humaines graves comme certains déficits immunitaires, cancers et maladies mentales. Rac1 est aussi la cible de nombreux facteurs de virulence bactériens [2]. L’étude d’un de ces facteurs, CNF1 (cytotoxic necrotizing factor 1), a permis à notre équipe de découvrir un nouveau mode de régulation des protéines Rho, notamment Rac1, par ubiquitinylation et dégradation protéasomique, et d’identifier certains des acteurs de cette régulation cellulaire, avec la découverte aujourd’hui de l’activité E3 ubiquitine ligase de HACE1 (HECT domain and ankyrin repeat containing E3 ubiquitin-protein ligase 1) vis-à-vis de Rac1 [3]. Ces observations posent la question de la relation entre cette activité biochimique de HACE1 et ses propriétés de suppresseur de tumeur.
Les transitions GTP/GDP des GTPases Ras
Rac1 comme les autres membres de la famille Rho (Rho/Rac/Cdc42), appartient à la superfamille des GTPases Ras [1]. Ce sont des protéines qui oscillent entre un état inactif et un état activé en réponse aux signaux transmis par des récepteurs situés à la membrane plasmique. Ces protéines lient la guanosine-5’-triphosphate (GTP) et catalysent son hydrolyse en GDP (Figure 1). Durant la transition GTP/GDP, deux domaines dits switch changent de conformation, offrant à ces GTPases, sous leur forme GTP, la propriété de fixer et d’activer des protéines effectrices. Les transitions GTP/GDP sont finement régulées par les facteurs d’échange (GEF) qui catalysent l’échange du GDP en GTP, et par les protéines GAP qui stimulent l’hydrolyse, spontanément faible, du GTP. Ce cycle de régulation temporelle est complété par une régulation spatiale. Sous forme GTP, Rac1 est localisée à la membrane, où s’exerce son activité de signalisation, alors qu’elle est séquestrée, sous forme inactive liée au GDP, dans le cytosol, en interaction avec le facteur GDI.
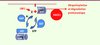 |
Figure 1. Régulation de Rac1 par HACE1. Représentation schématique du cycle spatiotemporel de régulation de Rac1 (gris), basé sur son activité GTPasique. Les facteurs régulateurs de l’échange et de l’hydrolyse du GTP en GDP sont indiqués en bleu. La forme activée de Rac1 (liée au GTP) est ciblée aux membranes (marron) auxquelles son groupement géranylgéranyl permet son ancrage. CNF1, en catalysant la déamidation de la glutamine 61 (Q à E), induit une forme dominante active de Rac1. Nos récents travaux établissent que la E3 ligase HACE1 (rouge) fixe la forme active de Rac1 et entraîne sa polyubiquitinylation à la membrane, conduisant à sa dégradation protéasomique. |
Contrôle des GTPases Rho et de Rac1 par ubiquitinylation et dégradation par le protéasome
Il est apparu, il y a quelques années, que les GTPases Rho et leur état d’activation étaient également contrôlés par un processus d’ubiquitinylation dégradative [4, 5]. Cette découverte repose sur l’étude de la toxine bactérienne CNF1, produite par des souches d’E. coli uropathogènes, agents responsables de cystites, de pyélonéphrites, et fréquemment de bactériémies [5]. CNF1 se fixe sur un récepteur à la surface des cellules pour pénétrer dans les compartiments intracellulaires à partir desquels elle transfère son domaine enzymatique dans le cytoplasme. Elle y catalyse la déamidation des GTPases Rho, dont Rac1. La déamidation spécifique d’une glutamine de Rac1 - en position 61 - en acide glutamique, confère à cette GTPase une activation constitutive et des propriétés classiques de mutant dominant positif. Cette modification de Rac1 par CNF1 facilite l’invasion des cellules de l’hôte par les bactéries [6]. De plus, l’étude de CNF1 a permis d’établir que les formes ainsi activées de Rac1 et des GTPases Rho subissent un processus d’ubiquitinylation entraînant leur adressage au protéasome et leur dégradation [4]. Loin d’être limité au modèle de la toxine CNF1, ce mécanisme d’ubiquitinylation et dégradation des GTPases Rho se produit également pour d’autres mutations activatrices ou lors de leur activation physiologique par les GEF. L’ubiquitinylation est une modification post-traductionnelle des protéines résultant de la fixation covalente de l’ubiquitine, un polypeptide de 8 kDa, à une lysine de la protéine cible [7]. Elle met en jeu une cascade de réactions d’activation et de transfert de l’ubiquitine réalisée séquentiellement par les enzymes E1 et E2 et qui aboutit à la fixation de l’ubiquitine sur la cible par une enzyme spécifique dite E3 ubiquitine ligase. La E3 ligase est capable de discriminer une cible et c’est donc elle qui assure la spécificité de la réaction d’ubiquitinylation d’une protéine [7]. D’autres molécules d’ubiquitine peuvent se fixer sur une des sept lysines de la première, constituant des enchaînements d’ubiquitines de longueur et de structure variées. L’enchaînement sur la lysine 48 (K48) de l’ubiquitine est un signal canonique de ciblage vers le protéasome. Ainsi, les formes actives des GTPases Rho subissent une polyubiquitinylation de type K48, notamment sur la lysine 147 de Rac1, entraînant leur dégradation par le protéasome [4].
L’E3 ubiquitine-ligase HACE1 catalyse l’ubiquitinylation de Rac1-GTP
Des études antérieures avaient permis d’identifier la protéine Smurf1, une E3 ubiquitine ligase de type HECT (homologous to E6-AP C-terminus), comme responsable de l’ubiquitinylation de la GTPase RhoA en réponse à son activation par CNF1 ou en aval de la signalisation par le TGF-bêta [4, 8]. Dans un nouveau travail publié en novembre 2011 dans Developmental Cell [3], nous avons établi un crible, basé sur l’activité de CNF1, pour déterminer le rôle potentiel des E3 ligases de type HECT dans l’ubiquitinylation et la dégradation de Rac1. Un crible fondé sur la déplétion systématique de chacune des 27 ligases à domaine HECT par la technique d’ARN interférence (ARNi) nous a permis d’identifier HACE1 comme étant essentielle à la dégradation protéasomique de Rac1 (mais non de RhoA) induite par CNF1 (Figure 1). Par des études d’interaction in vitro et de co-immunoprécipitation in vivo, nous avons établi que HACE1 s’associe préférentiellement à la forme active (liée au GTP) de Rac1. De plus HACE1 est colocalisée avec Rac1 à la périphérie de la cellule où se situe la forme active de cette GTPase. La réalisation de réactions d’ubiquitinylation in vitro utilisant des protéines recombinantes purifiées nous a permis de montrer que HACE1 catalyse cette réaction, avec une forte spécificité pour la forme active de Rac1. L’ensemble de ces résultats démontre que HACE1 fixe préférentiellement Rac1-GTP pour catalyser son ubiquitinylation. L’expression d’un mutant K48R de l’ubiquitine dans les cellules réduit fortement la formation de chaînes de polyubiquitines. Au total, ces données établissent l’activité E3 ubiquitine-ligase de HACE1 sur Rac1, et permettent l’identification d’une première cible cellulaire de HACE1.
Inactivation de HACE1 et progression tumorale
La déplétion de HACE1 par ARNi conduit à une augmentation des niveaux cellulaires de la forme active de Rac1 dans des cellules en culture et plus encore après stimulation par CNF1 (Figure 1). D’un point de vue physiologique et physiopathologique, nos résultats soulèvent la question des relations entre l’activité E3 ligase de HACE1 vis-à-vis de Rac1 et ses propriétés de suppresseur de tumeur [9]. En effet, le gène codant HACE1 chez l’homme se trouve dans la région chromosomique 6q21 qui possède une activité avérée de suppresseur de tumeur. Le gène HACE1 est inactivé par un mécanisme épigénétique dans de nombreuses tumeurs de Wilms ; il est également sous exprimé dans environ 50 % des tumeurs humaines et a été impliqué dans de multiples types de cancer chez l’homme. Chez la souris, l’inactivation de hace1 induit une susceptibilité accrue au développement de cancers de tous types au cours du vieillissement et coopère avec p53 dans la tumorigenèse [9]. Il semble que cet effet passe par une dérégulation de l’activité de la cycline D1, bien que celle-ci ne soit pas une cible directe de HACE1. Rac1 n’est pas à proprement parler une protéine oncogénique, mais sa contribution à divers aspects de la cancérogenèse, notamment à la prolifération cellulaire et à l’invasion tumorale, et son rôle essentiel dans la transformation par l’oncogène Ras, ont été largement mis en évidence [10, 11]. En particulier, la signalisation que contrôle Rac1 joue un rôle dans la dynamique des contacts intercellulaires, la migration et le cycle cellulaires. D’autre part, Rac1 a été impliquée in vivo dans divers types de tumeurs humaines soit parce qu’elle est surexprimée dans des tumeurs gastriques, testiculaires, mammaires, coliques, soit parce qu’elle est suractivée dans des tumeurs mammaires et prostatiques ainsi que dans les leucémies myéloïdes chroniques (LMC) [12]. Chez la souris, l’inactivation de Rac1 inhibe le développement de tumeurs bronchiques, coliques et cutanées induites par K-Ras. L’augmentation du niveau de Rac1-GTP qu’entraîne l’inactivation de HACE1 pourrait donc avoir des effets activateurs sur la prolifération et l’invasion tumorales.
Il reste à démontrer l’implication de cette régulation de Rac1 dans l’effet suppresseur de tumeur de HACE1. Cette voie de recherche est particulièrement intéressante au vu des récents travaux montrant que l’inactivation pharmacologique de Rac1 par le NSC23766 dans des formes de LMC résistantes à l’Imatinib réduit considérablement la progression tumorale [12]. Force est de constater que l’étude des pathogènes ouvre une fois encore de nouvelles voies de recherche en cancérologie.
Conflit d’intérêts
Les auteurs déclarent n’avoir aucun conflit d’intérêts concernant les données publiées dans cet article.
Remerciements
Notre recherche est financée par l’Inserm et le CNRS et par différents contrats ANR et ARC. Nous sommes aussi particulièrement reconnaissants à l’ARC et à la Ligue contre le cancer pour leur soutien continu de nos recherches par le financement de doctorants et post-doctorants.
Références
- Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol 2008 ; 9 : 690–701. [Google Scholar]
- Boquet P, Lemichez E. Bacterial virulence factors targeting Rho GTPases: parasitism or symbiosis? Trends Cell Biol 2003 ; 13 : 238–246. [CrossRef] [PubMed] [Google Scholar]
- Torrino S, Visvikis O, Doye A, et al. The E3 ubiquitin-ligase HACE1 catalyzes the ubiquitylation of active Rac1. Dev Cell 2011 ; 21 : 959–965. [CrossRef] [PubMed] [Google Scholar]
- Visvikis O, Maddugoda MP, Lemichez E. Direct modifications of Rho proteins: deconstructing GTPase regulation. Biol Cell 2010 ; 102 : 377–389. [CrossRef] [PubMed] [Google Scholar]
- Doye A, Mettouchi A, Bossis G, et al. CNF1 exploits the ubiquitin-proteasome machinery to restrict Rho GTPase activation for bacterial host cell invasion. Cell 2002 ; 111 : 553–564. [CrossRef] [PubMed] [Google Scholar]
- Visvikis O, Boyer L, Torrino S, et al. Escherichia coli producing CNF1 toxin hijacks tollip to trigger Rac1-dependent cell invasion. Traffic 2011 ; 12 : 579–590. [CrossRef] [PubMed] [Google Scholar]
- Weissman AM. Themes and variations on ubiquitylation. Nat Rev Mol Cell Biol 2001 ; 2 : 169–178. [CrossRef] [PubMed] [Google Scholar]
- Ozdamar B, Bose R, Barrios-Rodiles M, et al. Regulation of the polarity protein Par6 by TGFbeta receptors controls epithelial cell plasticity. Science 2005 ; 307 : 1603–1609. [CrossRef] [PubMed] [Google Scholar]
- Zhang L, Anglesio MS, O’Sullivan M, et al. The E3 ligase HACE1 is a critical chromosome 6q21 tumor suppressor involved in multiple cancers. Nat Med 2007 ; 13 : 1060–1069. [CrossRef] [PubMed] [Google Scholar]
- Mettouchi A, Klein S, Guo W, et al. Integrin-specific activation of Rac controls progression through the G(1) phase of the cell cycle. Mol Cell 2001 ; 8 : 115–127. [CrossRef] [PubMed] [Google Scholar]
- Mack NA, Whalley HJ, Castillo-Lluva S, et al. The diverse roles of Rac signaling in tumorigenesis. Cell Cycle 2011 ; 10 : 1571–1581. [CrossRef] [PubMed] [Google Scholar]
- Thomas EK, Cancelas JA, Chae HD, et al. Rac guanosine triphosphatases represent integrating molecular therapeutic targets for BCR-ABL-induced myeloproliferative disease. Cancer Cell 2007 ; 12 : 467–478. [CrossRef] [PubMed] [Google Scholar]
© 2012 médecine/sciences – Inserm / SRMS
Liste des figures
|
Figure 1. Régulation de Rac1 par HACE1. Représentation schématique du cycle spatiotemporel de régulation de Rac1 (gris), basé sur son activité GTPasique. Les facteurs régulateurs de l’échange et de l’hydrolyse du GTP en GDP sont indiqués en bleu. La forme activée de Rac1 (liée au GTP) est ciblée aux membranes (marron) auxquelles son groupement géranylgéranyl permet son ancrage. CNF1, en catalysant la déamidation de la glutamine 61 (Q à E), induit une forme dominante active de Rac1. Nos récents travaux établissent que la E3 ligase HACE1 (rouge) fixe la forme active de Rac1 et entraîne sa polyubiquitinylation à la membrane, conduisant à sa dégradation protéasomique. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.