")
")
| Issue |
Med Sci (Paris)
Volume 27, Number 8-9, Août–Septembre 2011
|
|
|---|---|---|
| Page(s) | 704 - 706 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/2011278010 | |
| Published online | 31 août 2011 | |
miR-122, un microARN « à tout fer »
miR-122, a microRNA gatekeeper of iron homeostasis
Inserm U1016, CNRS UMR 8104, Université Paris Descartes, Institut Cochin, 24, rue du Faubourg Saint-Jacques, 75014 Paris, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Les microARN (miARN) constituent une classe abondante de petits ARN non codants qui régulent l’expression des gènes à un niveau post-transcriptionnel et dont l’implication dans de nombreux processus biologiques fondamentaux - développement, différenciation et métabolisme - est désormais reconnue. Les miARN matures interagissent avec les régions 3’ non traduites (3’-UTR) des ARNm qu’ils contrôlent, provoquant ainsi soit leur dégradation, soit l’inhibition de leur traduction. Chaque miARN pouvant contrôler en moyenne 200 gènes [1], des calculs théoriques ont estimé que les miARN réguleraient jusqu’à 50 % de l’ensemble des gènes.
Le miARN-122 (miR-122) est spécifique du foie et très abondant, puisqu’il représente environ 70 % de l’ensemble des miARN dans ce tissu [2]. De nombreuses études suggèrent aujourd’hui que l’expression régulée de miR-122 est importante pour le maintien des fonctions hépatiques.
Ainsi, le blocage de miR-122 hépatique conduit-il à la diminution des taux de cholestérol plasmatique et de triglycérides chez les rongeurs et les primates. En outre, les souris chez lesquelles miR-122 a été inhibé sont résistantes au développement de la stéatose hépatique induite par un régime gras. Chez l’homme, l’expression de miR-122 est réprimée dans la cirrhose et le carcinome hépatocellulaire. Par ailleurs, de nombreuses études ont montré que miR-122 est crucial pour l’infection par le virus de l’hépatite C (VHC), la réplication du virus et la réponse au traitement par l’interféron. Enfin, il existe un lien fonctionnel entre miR-122 et les rythmes circadiens, la transcription de miR-122 étant elle-même sous contrôle circadien [3]. miR-122 apparaît donc comme un élément crucial du réseau de régulation de l’expression des gènes hépatiques et pourrait être nécessaire à la synchronisation des gènes à l’interface entre plusieurs voies métaboliques (revue [4]).
Contrôle de l’homéostasie du fer par l’hepcidine
Dans les premières études d’extinction de miR-122 in vivo utilisant des oligonucléotides anti-miR-122 (antagomir), Castoldi et al. [5] ont observé une augmentation des niveaux d’ARNm codant pour des activateurs de l’hormone du fer, l’hepcidine, suggérant à ces auteurs une implication de miR-122 dans le métabolisme du fer.
Le fer est vital pour les cellules. Il est en outre indispensable à la respiration, puisqu’il permet la fixation de l’oxygène à l’hémoglobine des globules rouges. Les déficits en fer entraînent des anémies marquées notamment par une oxygénation insuffisante des tissus. Au contraire, lorsqu’il se trouve en excès dans l’organisme, le fer devient toxique pour les cellules en favorisant l’apparition de radicaux libres détruisant progressivement le foie, le cœur, ou encore le pancréas, comme dans l’hémochromatose héréditaire [6, 8]. L’homéostasie du fer dans l’organisme est assurée par une hormone secrétée principalement par le foie, l’hepcidine. Cette hormone agit en régulant la concentration, le stockage et la distribution du fer dans l’organisme. Elle empêche l’export du fer des entérocytes, site de l’absorption intestinale du fer alimentaire, et des macrophages, site de recyclage du fer de l’hémoglobine, en dégradant l’exportateur du fer présent à la membrane de ces cellules, la ferroportine (revue [7]). La production d’hepcidine est directement liée aux besoins en fer de l’organisme. Ainsi, en présence de fer, pour éviter la toxicité du métal, l’expression de l’hepcidine hépatique est-elle augmentée afin de réduire l’apport de fer dans l’organisme. Cette activation est dépendante des protéines membranaires hémochromatosiques HFE, hémojuvéline (HJV) et du récepteur à la transferrine 2 (RTf2). Ces protéines forment vraisemblablement une « plateforme d’activation » interagissant avec le fer circulant (l’holotransferrine) permettant d’induire, en association avec d’autres protéines comme les récepteurs des BMP (bone morphogenic protein), l’expression de l’hepcidine. Une des voies majeures de signalisation à travers cette plateforme est la voie SMAD. L’interaction de BMP6, de ses récepteurs à activité tyrosine kinase (BMPR-I et BMPR-II) et du corécepteur HJV conduit à la phosphorylation des effecteurs cytoplasmiques SMAD1/5/8 et à la formation d’un complexe entre les SMAD1/5/8 phosphorylées et SMAD4 [9]. Ce complexe est alors transloqué dans le noyau où il active le promoteur du gène de l’hepcidine [6, 9].
Contrôle indirect du taux d’hepcidine par miR-122
Dans une publication récente, Castoldi et al. ont évalué un rôle potentiel de miR-122 dans le métabolisme du fer en analysant les conséquences d’une injection intrapéritonéale d’antagomiR-122 [5]. Trois semaines après l’injection, la quantité de miR-122 hépatique est considérablement diminuée (sans altération du profil d’expression des autres miR) et celle de ses cibles connues augmentée. Sur le plan hématologique, les souris traitées présentaient un déficit en fer systémique avec compensation médullaire (taille des globules rouges diminuée et réticulocytose accrue) ayant pour conséquence une diminution du fer hépatique.
Sur le plan de la régulation génique, l’observation intéressante était l’augmentation de l’expression hépatique des gènes du métabolisme du fer. Ainsi, l’expression des gènes codant les régulateurs de l’hepcidine, HFE, HJV et BMPR1a est induite après l’injection d’antagomir, ainsi que celle du gène de l’hepcidine lui même. Afin de déterminer si l’induction de ces gènes était directement liée à l’inhibition de miR-122 par l’antagomir, une analyse in silico de la présence des sites miR-122 dans leur ARNm a été réalisée. Seuls les messagers HJV, HFE et hepcidine présentent dans leur séquence 3’-UTR une région de liaison à miR-122. La fonction de cette région a été testée ex vivo en plaçant cette région en 3’ du gène luciférase. Si cette région est la cible de miR-122, on s’attend à ce que l’activité luciférase diminue après surexpression de miR-122 dans les cellules, par déstabilisation du transcrit luciférase-miR-122. C’est effectivement ce qui fut observé pour les séquences miR-122 issues d’HJV et de HFE alors que la séquence putative miR-122 de l’hepcidine, en revanche, n’affectait pas l’activité luciférase. Ainsi, seules les séquences 3’-UTR de HJV et HFE sont des cibles directes de miR-122.
Les souris ayant reçu l’antagomir ont une quantité de fer dans la rate augmentée. Les gènes de biosynthèse de l’hème, ainsi que celui codant pour RTf1, sont également augmentés dans ce tissu, suggérant une érythropoièse extra-médullaire compensatoire consécutive au déficit en fer systémique. En accord avec cette hypothèse, miR-451, qui est associé à la différentiation érythroïde et sous la dépendance du facteur de transcription érythroide GATA1, était également augmenté dans la rate de ces souris.
Ainsi, ce travail suggère que le rôle physiologique de miR-122, en réprimant directement l’expression HJV et HFE, pourrait être d’éviter une expression trop élevée d’hepcidine qui aurait pour conséquence une diminution de l’apport de fer entérocytaire et macrophagique (Figure 1). De fait, les auteurs montrent que la répression de miR-122 conduit à un déficit en fer plasmatique et hépatique.
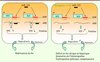 |
Figure 1 Rôle de miR-122 dans l’homéostasie du fer. MiR-122 est un répresseur de l’hepcidine, qui permet, par stabilisation de l’exportateur de fer, la ferroportine, un import de fer efficace dans l’organisme par absorption digestive et recyclage macrophagique accrus. L’extinction de miR-122 chez la souris par injection d’antagomir provoque l’augmentation des taux d’ARNm de HFE et d’HJV (deux cibles directes de miR-122) qui sont des activateurs de l’expression du gène de l’hepcidine. L’augmentation d’hepcidine a pour conséquence un déficit en fer systémique. |
La question importante est désormais de savoir si miR-122 est lui-même régulé par le fer et si les niveaux de miR-122 sont altérés dans les maladies liées au métabolisme du fer. En fait, les auteurs rapportent que chez les patients hémochromatosiques par déficit en hepcidine, les niveaux de miR-122 sont diminués. Cette diminution de miR-122 pourrait être, selon les auteurs, une réaction compensatoire visant à augmenter l’hepcidine pour limiter la surcharge en fer, mais cela reste encore à démontrer et d’autres rôles pourraient être attribués à miR-122.
En conclusion, la liste des processus physiopathologiques hépatiques impliquant miR-122 ne cesse de s’accroître puisqu’en dehors du métabolisme du cholestérol et des lipides, miR-122 semble également intervenir dans le métabolisme du fer. Connaissant les liens étroits entre métabolisme lipidique et métabolisme du fer, il est bien sûr tentant de spéculer que miR-122 pourrait être un élément-clé de l’interaction entre ces deux voies métaboliques.
Conflit d’intérêts
L’auteur déclare n’avoir aucun conflit d’intérêts concernant les données publiées dans cet article.
Références
- Krutzfeldt J, Stoffel M. MicroRNAs: a new class of regulatory genes affecting metabolism. Cell Metab 2006 ; 4 : 9-12. [CrossRef] [PubMed] [Google Scholar]
- Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of tissue-specific microRNAs from mouse. Curr Biol 2002 ; 12 : 735-739. [CrossRef] [PubMed] [Google Scholar]
- Gatfield D, Le Martelot G, Vejnar CE, et al. Integration of microRNA miR-122 in hepatic circadian gene expression. Genes Dev 2009 ; 23 : 1313-1326. [CrossRef] [PubMed] [Google Scholar]
- Girard M, Jacquemin E, Munnich A, Lyonnet S, Henrion-Caude A. miR-122, a paradigm for the role of microRNAs in the liver. J Hepatol 2008 ; 48 : 648-656. [CrossRef] [PubMed] [Google Scholar]
- Castoldi M, Vujic Spasic M, Altamura S, et al. The liver-specific microRNA miR-122 controls systemic iron homeostasis in mice. J Clin Invest 2011. [Google Scholar]
- Hentze MW, Muckenthaler MU, Galy B, Camaschella C. Two to tango : regulation of Mammalian iron metabolism. Cell 2010 ; 142 : 24-38. [CrossRef] [PubMed] [Google Scholar]
- Viatte L, Vaulont S. Hepcidin, the iron watcher. Biochimie 2009 ; 91 : 1223-1228. [CrossRef] [PubMed] [Google Scholar]
- Viatte I, Vaulont, S. Prévenir et guérir les surcharges en fer, les espoirs de l’hepcidine. Med Sci (Paris) 2006 ; 22 : 696-718. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Roth MP, Coppin H. BMP6, un acteur clé dans la régulation du métabolisme du fer. Med Sci (Paris) 2009 ; 25 : 678-680. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
© 2011 médecine/sciences – Inserm / SRMS
Liste des figures
|
Figure 1 Rôle de miR-122 dans l’homéostasie du fer. MiR-122 est un répresseur de l’hepcidine, qui permet, par stabilisation de l’exportateur de fer, la ferroportine, un import de fer efficace dans l’organisme par absorption digestive et recyclage macrophagique accrus. L’extinction de miR-122 chez la souris par injection d’antagomir provoque l’augmentation des taux d’ARNm de HFE et d’HJV (deux cibles directes de miR-122) qui sont des activateurs de l’expression du gène de l’hepcidine. L’augmentation d’hepcidine a pour conséquence un déficit en fer systémique. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.