")
")
| Issue |
Med Sci (Paris)
Volume 27, Number 5, Mai 2011
Représentation en sciences du vivant
|
|
|---|---|---|
| Page(s) | 547 - 552 | |
| Section | Forum | |
| DOI | https://doi.org/10.1051/medsci/2011275022 | |
| Published online | 25 mai 2011 | |
Microscopies cellulaires à l’échelle de la molécule individuelle
Représentation en sciences du vivant (8)
Imaging of single molecules in live cells
1
Institut de biologie de l’École normale supérieure, 46, rue d’Ulm, 75005 Paris
2
Laboratoire Kastler Brossel, département de physique, 46, rue d’Ulm, 75005 Paris
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
La possibilité de détecter des molécules uniques dans une cellule offre des modalités nouvelles pour l’imagerie biologique. Ainsi, des structures macromoléculaires peuvent être observées au sein de la cellule avec une résolution de 10 à 50 nm, bien inférieure à la limite de diffraction de la lumière (250 nm). L’enregistrement des trajectoires de protéines individuelles, qu’elles soient membranaires ou intracellulaires, permet également d’analyser les processus physiques et biochimiques qui contribuent à leur mouvement dans la cellule. À l’avenir, ces outils d’imagerie ultrasensible devraient largement contribuer à notre compréhension de l’organisation du vivant.
Abstract
Progress in optical microscopy, combined to the emergence of new fluorescent probes and advanced instrumentation, now permits the imaging of single molecules in fixed and live cells. This extreme detection sensitivity has opened new modalities in cellular imaging. On the one hand, optical images with an unprecedented resolution in the 10-50 nm range, well below the diffraction limit of light, can be recorded. These super-resolution images give new insights into the properties of cellular structures. On the other hand, proteins, either in the membrane or intracellular, can be tracked in live cells and in physiological conditions. Their individual trajectories provide invaluable information on the molecular interactions that control their dynamics and their spatial organization. Single molecule imaging is rapidly becoming a unique tool to understand the biochemical and biophysical processes that determine the properties of molecular assemblies in a cellular context.
© 2011 médecine/sciences – Inserm / SRMS
Au cours des quinze dernières années, l’imagerie optique a largement renouvelé notre compréhension de l’organisation et du fonctionnement de la cellule. Bien loin d’être des structures à l’organisation statique, les cellules vivantes sont des systèmes au sein desquels des assemblages moléculaires se forment et se défont en permanence, au gré des besoins fonctionnels [1]. Déchiffrer la nature des processus biophysiques et biochimiques qui contribuent à cette architecture dynamique et à sa régulation est ainsi devenu un enjeu important en biologie cellulaire.
Notre perception nouvelle de l’organisation cellulaire est en grande partie due aux progrès des techniques de microscopie utilisées pour observer le vivant. L’avancée la plus importante a incontestablement été le développement des protéines fluorescentes (récompensé en 2008 par l’attribution du prix Nobel de chimie à S. Imamura, M. Chalfie et R. Tsien) [2, 3]. L’utilisation de la green fluorescent protein (GFP) et de ses variantes permet en effet de localiser des structures spécifiques à l’échelle de la cellule ou de l’organisme vivant et d’en suivre la dynamique spatiale et temporelle [1]. Des améliorations instrumentales ont également contribué à accroître significativement les performances des microscopes de laboratoire. Ainsi les détecteurs (tels que les caméras CCD [charge-coupled device] intensifiées) permettent aujourd’hui l’acquisition de signaux optiques à des cadences élevées (jusqu’à plusieurs centaines d’images par seconde) et avec des sensibilités atteignant celle du photon individuel. Associés à la puissance de calcul des processeurs actuels, les microscopes modernes permettent l’acquisition et le traitement de quantités considérables de données. À ce titre, ils sont des outils essentiels pour la recherche biomédicale, fondamentale et appliquée.
La limite de détection optique en imagerie cellulaire a longtemps été celle de l’organite ou de petits agrégats moléculaires. Toutefois, au cours des dix dernières années, la combinaison de nouvelles techniques de microscopie, de sondes biologiques performantes et de traitement d’images a permis d’atteindre la sensibilité ultime, celle de la molécule unique. Nous discutons ici deux champs d’application particulièrement importants de l’imagerie de molécules individuelles en biologie cellulaire : la microscopie haute résolution et le suivi dynamique de molécules uniques.
Microscopie optique à haute résolution, ou comment vaincre la limite de la diffraction
La limite de la diffraction
Une question importante en microscopie est celle de la résolution spatiale dans l’image, c’est-à-dire la distance minimale pour laquelle on peut distinguer deux objets [4]. Depuis la fin du xix
e siècle, on sait que les lois de l’optique imposent une limite inférieure à cette résolution. Cette limite d, appelée limite de la diffraction ou loi d’Abbé, est égale à :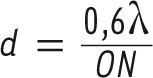
où λ est la longueur d’onde de la lumière avec laquelle l’échantillon est observé et ON est l’ouverture numérique du dispositif optique avec lequel la lumière est collectée (Figure 1). En biologie, où de la lumière visible est utilisée en général (λ comprise entre 400 et 700 nm) et où les meilleurs objectifs de microscope ont une ON de l’ordre de 1,4, d est au mieux de l’ordre de 200 nm, intermédiaire entre la taille d’assemblages supramoléculaires (10 à 100 nm) et celle d’une cellule (1 à 40 microns). En d’autres termes, la microscopie optique est très efficace pour nous renseigner sur l’organisation à l’échelle cellulaire mais bien moins adaptée pour nous informer sur des structures moléculaires.
 |
Figure 1. Microscopie de super-résolution par photoactivation. A.Limite de la diffraction en microscopie. Des sources de lumière ponctuelle apparaissent comme des taches étendues de largeur d (courbe grise). Tant que leur séparation reste supérieure (à gauche) ou égale (au centre) à d, ces points peuvent être résolus individuellement. En revanche, lorsqu’ils sont situés à une distance plus faible (à droite), leurs signaux se superposent (en rouge) et on ne peut plus les distinguer. B. Principe de la microscopie haute résolution avec des molécules photoconvertibles. Lorsqu’elles émettent dans le vert, elles ne sont pas distinguables. Si on en active un petit nombre, on peut mesurer les spots de fluorescence de molécules individuelles (en rouge). En déterminant le centre de ces spots, la distribution spatiale des molécules est reconstruite point à point. C. Exemple d’image haute résolution obtenue à partir d’un marquage du cytosquelette d’actine dans des cellules nerveuses en culture. L’image conventionnelle est présentée en vert. Après photoactivation, on obtient quelques spots dans le champ de vue (au centre) que l’on peut localiser avec une précision d’environ 25 nm. En répétant ce processus, une image haute résolution est obtenue (à droite). |
Pendant plus d’un siècle, la limite de la diffraction a été considérée comme inviolable, et seule la microscopie électronique (pour laquelle λ est bien inférieure) semblait pouvoir offrir des images avec une résolution atteignant ou dépassant celle du nanomètre. Toutefois, les dernières années ont été marquées par l’émergence de nouvelles méthodes optiques permettant de contourner cette limite [5, 6]. Ces méthodes peuvent être schématiquement scindées selon deux approches. Une première approche, que nous ne détaillons pas ici, utilise des faisceaux d’excitation structurés spatialement. C’est le cas de la microscopie STED (stimulated-emission-depletion ou déplétion par émission stimulée) inventée par Stefan Hell [5] ou de l’illumination structurée développée par Mats Gustaffson et John Sedat [7] qui permettent aujourd’hui d’obtenir des images d’échantillons biologiques vivants avec une résolution entre 40 et 100 nm environ. Une deuxième méthode, introduite en 2006 par les groupes américains d’Eric Betzig, Xiaowei Zhuang et Samuel Hess [8] et détaillée ci-dessous, utilise la microscopie en champ large conventionnelle mais tire parti de la possibilité de détecter et d’analyser le signal de molécules uniques.
La microscopie par photoactivation de molécules individuelles
Ordinairement, dans un microscope en champ large, une grande zone de l’échantillon est excitée. Une image sur une caméra est alors formée par la collection simultanée de la fluorescence de toutes les molécules présentes dans la zone. À cause de la diffraction, chaque molécule apparaît comme une tache d’extension d, ce qui détermine la résolution de l’image (Figure 1).
L’astuce pour augmenter la résolution consiste à n’allumer qu’une petite fraction des molécules de l’échantillon, de façon à ce que leur distance relative soit supérieure à d. Dans ce cas, chaque tache dans l’image ne correspond qu’à une seule molécule, que l’on peut localiser en déterminant le centre de la tache de diffraction (Figure 1). Il s’avère, et c’est là un point crucial, que la précision σ de localisation est bien meilleure que d. En fait, σ varie comme d/√N où N est le nombre de photons dans la tache de fluorescence, et atteint des valeurs comprises entre 10 et 40 nm dans des conditions typiques d’imagerie d’une cellule. Cette précision peut même être subnanométrique dans l’analyse d’échantillons in vitro.
Pour obtenir l’image complète de l’échantillon, il faut donc allumer quelques molécules, les localiser par analyse d’image puis les éteindre avant de recommencer avec une autre fraction des molécules. En répétant un grand nombre de fois cette séquence d’allumage-localisalisation-extinction, on reconstruit molécule par molécule une image dont la résolution est déterminée par la précision de localisation σ. Ce type de microscopie, décrite parfois comme du pointillisme moléculaire par analogie avec la technique de peinture du même nom, est désigné dans la littérature sous les acronymes PALM (photoactivated localization microscopy) [8, 9] ou STORM (stochastic optical reconstruction microscopy) [10].
L’élément-clé est de disposer de sondes fluorescentes que l’on peut allumer puis éteindre à volonté. Plusieurs types de marqueurs - protéines de fusion ou colorants organiques - permettent aujourd’hui cela [11]. Les protéines photoconvertibles sont sans doute l’un des systèmes les plus intéressants. Ces protéines, qui émettent naturellement à une longueur d’onde (en général dans le vert), changent de couleur et émettent à une autre (dans le rouge) après activation par de la lumière violette. Ainsi lorsque la fluorescence verte est détectée, une image de résolution standard est obtenue car toutes les molécules émettent en même temps. Après un très bref flash de lumière violette, seules quelques molécules sont converties et peuvent être détectées de manière isolée en collectant la fluorescence rouge. Après quelques instants d’illumination, ces molécules activées s’éteignent par photodestruction. Le grand avantage de ces protéines est que, comme pour la GFP, elles peuvent être fusionnées à des gènes d’intérêt, permettant ainsi de regarder l’organisation de protéines spécifiques dans des cellules vivantes avec une résolution nanométrique (Figure 1).
Suivi dynamique de molécules uniques
Une deuxième application de l’imagerie de molécules uniques est le suivi dynamique dans la cellule. Dans ce cas est enregistré dans une séquence d’images le signal d’une molécule individuelle marquée par une sonde fluorescente. De même que dans la microscopie PALM ou STORM, la molécule est localisée dans chaque image avec une précision σ inférieure à la limite de diffraction. L’analyse de la trajectoire, reconstruite à partir des positions successives au cours de la séquence, donne accès à des paramètres importants qui contrôlent la dynamique spatiale, tels que la vitesse de transport ou le coefficient de diffusion. Tout aussi importante est l’analyse d’interactions moléculaires lorsque la molécule s’immobilise sur un site particulier. Deux exemples permettent d’illustrer l’intérêt biologique de ces expériences de suivi de molécules uniques.
Diffusion membranaire des récepteurs des neurotransmetteurs
Comprendre les mécanismes par lesquels des récepteurs de neurotransmetteurs atteignent et régulent leur localisation synaptique est un problème central pour la biologie des neurones. Depuis une dizaine d’années, un ensemble d’expériences sur récepteurs uniques ont permis de mettre en évidence des processus nouveaux, difficiles à identifier par des mesures sur des populations de récepteurs [12]. En attachant un marqueur fluorescent aux récepteurs du glutamate AMPA et NMDA, aux récepteurs GABA (qui fixent l’acide gamma-aminobutyrique) ou au récepteur de la glycine, et en suivant leurs mouvements, on observe que ces récepteurs, loin d’être statiques à la synapse, sont animés d’une agitation incessante. Ils diffusent dans la membrane extrasynaptique, rentrent dans la synapse mais bien souvent en sortent rapidement avant de diffuser et d’atteindre une autre synapse (Figure 2). Cette dynamique membranaire, qui s’ajoute aux mécanismes d’endocytose/exocytose, contribue à la distribution spatiale des récepteurs et ainsi à la régulation du nombre de récepteurs à la synapse. Au-delà de la description qualitative des mouvements, les expériences de suivi ont permis de mesurer précisément les propriétés de diffusion dans la membrane mais aussi les temps de résidence à la synapse. Ces derniers traduisent la force des interactions moléculaires du récepteur avec les protéines d’ancrage qui le stabilisent (au moins transitoirement) au niveau des sites synaptiques.
 |
Figure 2. Suivi dynamique d’un récepteur de la glycine. A. Le récepteur est marqué par une nanoparticule fluorescente (vert). Les protéines d’échafaudage à la membrane post-synaptique sont visualisées par un marqueur (rouge). B1 à B6. Mouvement du récepteur (vert) dans la membrane neuronale. Il diffuse dans la membrane avant d’atteindre le site synaptique (B4) où il ne reste que transitoirement lié. C. Reconstruction de la trajectoire avec des phases extrasynaptique (bleu) et synaptique (vert). Adapté de [17]. |
Dynamique nucléaire des facteurs de transcription
Un effort important est fait actuellement pour accéder à la dynamique de molécules uniques dans des compartiments intracellulaires. Ces expériences, bien plus délicates que pour les protéines membranaires, suggèrent également une grande complexité des comportements moléculaires. Des mesures récentes de la dynamique nucléaire du facteur de transcription pTEFb illustrent cela (Figure 3). Un facteur de transcription est une protéine qui contrôle l’expression génique en se fixant à des séquences cibles dans le génome. Lorsqu’ils sont observés individuellement, ces facteurs de transcription diffusent rapidement dans le noyau, si rapidement que le signal de fluorescence apparaît comme une tache floue à l’échelle du temps d’acquisition de l’image. Occasionnellement, le signal change et ressemble à la tache de diffraction bien définie d’une molécule quasi immobile. Ces événements correspondent probablement à l’interaction transitoire du facteur de transcription avec la chromatine. En mesurant la durée de ces événements d’interaction, on peut donc déterminer l’affinité de la protéine pour des sites le long de l’ADN et ainsi étudier les paramètres qui contrôlent la cinétique de la régulation de l’expression génique.
 |
Figure 3. Dynamique nucléaire des facteurs de transcriptions. A1, A2. Principe du suivi dynamique par photoactivation. Au sein d’une population de molécules (points bleus), une petite fraction d’entre elles (rouge) est photoactivée, avec une densité suffisamment faible pour qu’on puisse les suivre individuellement. B1 à B4. Représentation tridimensionnelle de l’intensité de fluorescence à différents instants. Les spots bien définis correspondent à des molécules quasi immobiles à l’échelle de temps de l’acquisition (10 ms). Les taches plus étalées (images B2 et B4) indiquent que la molécule bouge rapidement durant l’acquisition. C. Exemple de trajectoires : molécule quasi immobile (rouge), diffusion lente (bleu) ou rapide (gris). La barre d’échelle grise correspond à 1 µm. La trajectoire grise suggère aussi que la molécule alterne entre un mouvement rapide et une phase ralentie correspondant par exemple à une interaction transitoire avec des partenaires protéiques ou avec la chromatine. L’insert est un zoom sur la trajectoire quasi immobile (barre d’échelle : 50 nm). |
Perspectives
Enjeux techniques et méthodologiques
Moins de dix ans après les premières mesures de suivi de molécules uniques et cinq ans après les premières images PALM/STORM haute résolution, l’imagerie de molécules uniques s’est imposée comme un outil puissant pour l’investigation du vivant. Toutefois, de nombreux défis techniques et méthodologiques restent à résoudre, tels que celui d’obtenir des trajectoires tridimensionnelles, de gagner en résolution spatiale et temporelle ou d’étendre les mesures de molécules uniques à des systèmes multicellulaires tels que des tissus, des tranches de cerveau ou des petits animaux.
Une des clés est certainement d’améliorer les propriétés des sondes fluorescentes. Ainsi, des sondes plus brillantes permettraient d’augmenter le nombre N de photons détectés et donc la précision de localisation σ dans les images haute résolution. Cela permettrait d’atteindre, ou au moins de se rapprocher de ce qui est le Graal actuel de la microscopie optique sous la limite de la diffraction, c’est-à-dire une résolution de 5 nm, au lieu de 10-40 nm actuellement. Avec une telle résolution, on accède à une échelle véritablement moléculaire et on pourrait non seulement visualiser de petits assemblages moléculaires mais aussi sonder leur organisation interne directement dans la cellule.
Pour les expériences de suivi de molécules, c’est la stabilité de la sonde qui est cruciale, afin d’enregistrer des trajectoires longues, de durée compatible avec celle des processus biologiques d’intérêt. Comme la durée d’émission des sondes organiques est souvent limitée à quelques secondes par les phénomènes de photodestruction, un effort important - et fructueux - a été mené pour utiliser des marqueurs inorganiques tels que des nanocristaux semi-conducteurs [13]. Ces nanoparticules, brillantes et photostables, ont déjà permis des expériences de suivi de protéines membranaires individuelles pendant plusieurs minutes (Figure 2). Par ailleurs, leurs propriétés spectrales d’absorption et d’émission facilitent grandement les mesures multi-couleurs nécessaires pour corréler le mouvement de la biomolécule marquée à son environnement moléculaire. D’autres nano-objets, tels que les nanoparticules d’or [14] ou les nanodiamants [15], offrent des alternatives possibles aux nanoparticules conductrices pour le suivi de molécules. Toutefois, l’utilisation de nanoparticules, quelle que soit leur nature physique, soulève toujours des difficultés particulières par rapport aux molécules organiques. Il est notamment indispensable de contrôler précisément leurs propriétés colloïdales pour permettre leur solubilisation, leur fonctionnalisation et leur ciblage vers des molécules membranaires ou intracellulaires. De plus, la taille des nanoparticules solubilisées peut atteindre 25 nm, celle d’un gros complexe macromoléculaire. Un enjeu important pour l’utilisation de nanoparticules comme sondes biologiques est donc de réduire leurs dimensions tout en conservant leurs caractéristiques optiques, afin de se rapprocher des échelles moléculaires et de minimiser les perturbations potentielles sur la dynamique et l’activité de la cible.
Que cela soit pour l’amélioration des sondes organiques ou inorganiques, les problèmes à résoudre sont par essence pluridisciplinaires et requièrent donc des solutions qui combinent des techniques empruntant à l’optique, la physicochimie, la biochimie et la biologie moléculaire et cellulaire.
Vers la biochimie in situ et la biologie des systèmes
Un des aspects les plus fascinants des mesures de suivi est la possibilité de sonder des interactions moléculaires directement dans des cellules vivantes. En mesurant les temps d’association et de dissociation entre un récepteur et des protéines d’ancrage ou entre un facteur de transcription et l’ADN, on accède à des paramètres cinétiques in situ. Ces expériences dans la cellule tiennent compte de nombreux facteurs difficiles à reproduire dans des analyses biochimiques en tube à essai tels que l’environnement chimique local, la présence de compétiteurs multiples, les inhomogénéités de concentration ou la « dimensionnalité » du milieu.
Par ailleurs, de nombreuses expériences en molécules uniques ont montré que les paramètres cinétiques, tels que des taux de dissociation ou des coefficients de diffusion, varient considérablement d’une molécule à l’autre. Cette variabilité s’explique en partie par la nature stochastique des interactions entre biomolécules, mais elle peut aussi être le reflet d’une propriété spécifique de l’organisation cellulaire. De même que les expériences en cellules uniques ont brisé le mythe de la cellule « typique », les mesures sur molécules individuelles remettent souvent en cause l’idée d’un comportement moléculaire moyen. À terme, un défi pour la modélisation du vivant est donc d’intégrer les propriétés moléculaires telles qu’elles émergent des expériences sur molécules individuelles dans une description de la dynamique fonctionnelle à l’échelle cellulaire. En d’autres termes, comment faire le lien entre la molécule et le système ?
Conclusion
Aujourd’hui, grâce à la détection de molécules uniques, on est capable de former des images à des résolutions nanométriques, de suivre des molécules en pleine activité dans la cellule et, dans des expériences que nous n’avons pas décrites ici, de compter des molécules [16] au sein de complexes multiprotéiques. On dispose donc d’une palette d’outils permettant d’analyser la structure, la dynamique et la composition d’assemblages moléculaires dans des contextes physiologiques. À terme, l’imagerie de molécules individuelles devrait non seulement permettre d’apporter des réponses à des questions biologiques précises mais aussi de soulever des points conceptuels nouveaux sur la dynamique et l’organisation du vivant. ‡
Conflit d’intérêts
Les auteurs déclarent avoir des liens durables (contrat de recherche) avec l’entreprise Nikon France.
Remerciements
Les auteurs remercient Lana Bosanac, Ibrahim Cissé, Marie Ehrensperger, Vincent Récamier, Christian Specht et les autres membres des groupes « Optique et biologie » et « Imagerie de la machinerie transcriptionnelle » pour leur contribution aux données, ainsi qu’Antoine Triller et Christophe Zimmer pour des discussions fructueuses.
Références
- Coscoy C, Amblard F. Le temps, sculpteur de la cellule ? Med Sci (Paris) 2011 ; 27 : 452-431. [Google Scholar]
- Tsien RY. Building and breeding molecules to spy on cells and tumors. FEBS Lett 2005 ; 579 : 927-932. [CrossRef] [PubMed] [Google Scholar]
- Salamero J. Prix Nobel de chimie 2008 (Osumo Shimomura, Martin Chalfie et Roger Y. Tsien). La « révolution verte » est en marche. Med Sci (Paris) 2008 ; 24 : 987-988. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Hecht E. Optique. Paris : Pearson Education, 2005 : 698 p. [Google Scholar]
- Hell SW. Far-field optical nanoscopy. Science 2007 ; 316 : 1153-1158. [CrossRef] [PubMed] [Google Scholar]
- Huang B, Bates M, Zhuang X. Super-resolution fluorescence microscopy. Annu Rev Biochem 2009 ; 78 : 993-1016. [CrossRef] [PubMed] [Google Scholar]
- Gustafsson MG, Shao L, Carlton PM, et al. Three-dimensional resolution doubling in wide-field fluorescence microscopy by structured illumination. Biophys J 2008 ; 94 : 4957-4970. [CrossRef] [PubMed] [Google Scholar]
- Betzig E, Patterson GH, Sougrat R, et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006 ; 313 : 1642-1645. [CrossRef] [PubMed] [Google Scholar]
- Hess ST, Girirajan TP, Mason MD. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys J 2006 ; 91 : 4258-4272. [CrossRef] [PubMed] [Google Scholar]
- Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat Methods 2006 ; 3 : 793-795. [Google Scholar]
- Fernandez-Suarez M, Ting AY. Fluorescent probes for super-resolution imaging in living cells. Nat Rev Mol Cell Biol 2008 ; 9 : 929-943. [CrossRef] [PubMed] [Google Scholar]
- Triller A, Choquet D. New concepts in synaptic biology derived from single-molecule imaging. Neuron 2008 ; 59 : 359-374. [CrossRef] [PubMed] [Google Scholar]
- Pinaud F, Clarke S, Sittner A, Dahan M. Probing cellular events, one quantum dot at a time. Nat Methods 2010 ; 7 : 275-285. [CrossRef] [PubMed] [Google Scholar]
- Lasne D, Blab GA, Berciaud S, et al. Single nanoparticle photothermal tracking (SNaPT) of 5-nm gold beads in live cells. Biophys J 2006 ; 91 : 4598-4604. [Google Scholar]
- Faklaris O, Garrot D, Joshi V, et al. Detection of single photoluminescent diamond nanoparticles in cells and study of the internalization pathway. Small 2008 ; 4 : 2236-2239. [CrossRef] [PubMed] [Google Scholar]
- Ulbrich MH, Isacoff EY. Subunit counting in membrane-bound proteins. Nat Methods 2007 ; 4 : 319-321. [PubMed] [Google Scholar]
- Ehrensperger MV, Hanus C, Vannier C, et al. Multiple association states between glycine receptors and gephyrin identified by SPT analysis. Biophys J 2007 ; 92 : 3706-3718. [CrossRef] [PubMed] [Google Scholar]
Liste des figures
|
Figure 1. Microscopie de super-résolution par photoactivation. A.Limite de la diffraction en microscopie. Des sources de lumière ponctuelle apparaissent comme des taches étendues de largeur d (courbe grise). Tant que leur séparation reste supérieure (à gauche) ou égale (au centre) à d, ces points peuvent être résolus individuellement. En revanche, lorsqu’ils sont situés à une distance plus faible (à droite), leurs signaux se superposent (en rouge) et on ne peut plus les distinguer. B. Principe de la microscopie haute résolution avec des molécules photoconvertibles. Lorsqu’elles émettent dans le vert, elles ne sont pas distinguables. Si on en active un petit nombre, on peut mesurer les spots de fluorescence de molécules individuelles (en rouge). En déterminant le centre de ces spots, la distribution spatiale des molécules est reconstruite point à point. C. Exemple d’image haute résolution obtenue à partir d’un marquage du cytosquelette d’actine dans des cellules nerveuses en culture. L’image conventionnelle est présentée en vert. Après photoactivation, on obtient quelques spots dans le champ de vue (au centre) que l’on peut localiser avec une précision d’environ 25 nm. En répétant ce processus, une image haute résolution est obtenue (à droite). |
| Dans le texte | |
|
Figure 2. Suivi dynamique d’un récepteur de la glycine. A. Le récepteur est marqué par une nanoparticule fluorescente (vert). Les protéines d’échafaudage à la membrane post-synaptique sont visualisées par un marqueur (rouge). B1 à B6. Mouvement du récepteur (vert) dans la membrane neuronale. Il diffuse dans la membrane avant d’atteindre le site synaptique (B4) où il ne reste que transitoirement lié. C. Reconstruction de la trajectoire avec des phases extrasynaptique (bleu) et synaptique (vert). Adapté de [17]. |
| Dans le texte | |
|
Figure 3. Dynamique nucléaire des facteurs de transcriptions. A1, A2. Principe du suivi dynamique par photoactivation. Au sein d’une population de molécules (points bleus), une petite fraction d’entre elles (rouge) est photoactivée, avec une densité suffisamment faible pour qu’on puisse les suivre individuellement. B1 à B4. Représentation tridimensionnelle de l’intensité de fluorescence à différents instants. Les spots bien définis correspondent à des molécules quasi immobiles à l’échelle de temps de l’acquisition (10 ms). Les taches plus étalées (images B2 et B4) indiquent que la molécule bouge rapidement durant l’acquisition. C. Exemple de trajectoires : molécule quasi immobile (rouge), diffusion lente (bleu) ou rapide (gris). La barre d’échelle grise correspond à 1 µm. La trajectoire grise suggère aussi que la molécule alterne entre un mouvement rapide et une phase ralentie correspondant par exemple à une interaction transitoire avec des partenaires protéiques ou avec la chromatine. L’insert est un zoom sur la trajectoire quasi immobile (barre d’échelle : 50 nm). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.