")
")
| Issue |
Med Sci (Paris)
Volume 27, Number 5, Mai 2011
|
|
|---|---|---|
| Page(s) | 501 - 507 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/2011275015 | |
| Published online | 25 May 2011 | |
La protéine C activée
Une protéine à l’interface de l’inflammation et de la coagulation
Activated protein C
A protein at the crossroads between coagulation and inflammation
1
AP-HP, Service de réanimation médicale, Hôpital Ambroise Paré, 9 av Charles de Gaulle, 92100 Boulogne Billancourt
2
EA4531, Faculté de Pharmacie, Université Paris-Sud, 5 rue Jean-Baptiste Clément, 92296 Chatenay-Malabry, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
Le sepsis est défini comme une réponse systémique de l’organisme à une infection. Au plan physiopathologique, la réponse de l’hôte à l’infection est caractérisée, d’une part par une réponse inflammatoire intense, et d’autre part par une activation de la coagulation et une inhibition de la fibrinolyse, ces processus étant étroitement imbriqués. Dans un domaine où la mortalité reste très élevée, une approche nouvelle a démontré son efficacité en termes d’amélioration du pronostic vital. Il s’agit d’un traitement par administration de protéine C activée, un inhibiteur physiologique de la coagulation doué de propriétés cytoprotectrices. La protéine C activée appartient à un système qui fait intervenir des protéines plasmatiques et des récepteurs présents en particulier à la surface des cellules endothéliales. En plus de son effet largement documenté sur la coagulation et la fibrinolyse, elle possède des activités anti-inflammatoire, anti-apoptotique mais aussi anti-histone. En effet, une récente étude menée sur les effets cytoprotecteurs de la protéine C activée a montré que des histones extracellulaires sont libérées au cours du sepsis sévère et semblent participer à la physiopathologie de ce dernier. Ces histones sembleraient être une nouvelle cible de la protéine C activée.
Abstract
Sepsis is defined as a systemic response to infection, characterized by an intense inflammatory response linked to coagulation activation and fibrinolysis inhibition, two processes which are intimately associated. In a field where mortality remains very high, administration of activated protein C, a physiological coagulation inhibitor with cytoprotective properties, has demonstrated its effectiveness and was able to reduce mortality. Protein C belongs to a system that involves plasma proteins and endothelial cell receptors. In addition to well documented effects on coagulation and fibrinolysis, activated protein C exhibits anti-inflammatory, anti-apoptotic but also anti-histone activities. Indeed, a recent study focusing on the cytoprotective effects of activated protein C showed that extracellular histones are released during severe sepsis and may participate in the pathophysiology of severe sepsis. These histones appear to be new targets of activated protein C.
© 2011 médecine/sciences – Inserm / SRMS
La protéine C activée : un anticoagulant physiologique
La coagulation met en jeu des protéines circulantes inactives qui acquièrent successivement une activité catalytique de type sérine protéase. Le déclenchement de cette cascade se fait grâce au facteur tissulaire (FT) : l’expression de cette protéine membranaire est normalement réprimée dans les cellules du sang et les cellules endothéliales vasculaires, mais une blessure vasculaire entraîne l’exposition du FT présent dans le sous-endothélium. Cette exposition de FT permet d’enclencher la coagulation qui, après une phase d’amplification impliquant différents cofacteurs dont les facteurs Va et VIIIa, aboutit à la génération de concentrations élevées de thrombine capable de transformer le fibrinogène soluble en un réseau de fibrine polymérisée.
L’activation de la coagulation observée au cours du sepsis est due à l’induction de FT à la surface des monocytes et des cellules endothéliales exposés à des composants bactériens (Figure 1). Cette réaction de l’organisme a pour objectif de former un réseau de fibrine destiné à emprisonner et éliminer les composants bactériens responsables de l’infection.
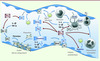 |
Figure 1. Effets de la protéine C activée au cours du sepsis. Les traits se terminant par une flèche correspondent à un effet activateur, les traits se terminant par un rond à un effet inhibiteur. Les traits rouges correspondent aux effets de la protéine C activée. PC : protéine C ; PCa : PC activée ; FT : facteur tissulaire ; TM : thrombomoduline ; EPCR : récepteur endothélial de la protéine C ; tPA : activateur tissulaire du plasminogène ; PAI-1 : inhibiteur de l’activateur du plasminogène ; H3 : histone extracellulaire 3 ; VCAM : vascular cell adhesion molecule ; ICAM : intercellular cell adhesion molecule ; IL-6, IL-1β : interleukines ; TNF-α : tumor necrosis factor. |
Le système de la coagulation est contrôlé par plusieurs mécanismes inhibiteurs dont le système de la protéine C (PC). La PC est une protéine dont la synthèse dépend de la vitamine K [42]. Physiologiquement, elle circule très majoritairement sous la forme d’un zymogène inactif et acquiert une activité sérine protéase une fois clivée par la thrombine. C’est donc la thrombine formée lors de l’activation de la coagulation qui va limiter sa propre génération en activant ce système anticoagulant naturel. L’activation de la PC par la thrombine est une réaction extrêmement lente qui est considérablement accélérée (20 000 fois) en présence de thrombomoduline (TM), un récepteur endothélial qui agit comme un cofacteur dans ce système [1]. Cette activation de la PC par le complexe thrombine/TM s’accroît encore d’un facteur 20 lorsque la PC est présentée par un second récepteur endothélial, l’endothelial cell protein C receptor (EPCR) [2] (Figure 2). La TM et l’EPCR, présents à la surface des cellules endothéliales et des monocytes, peuvent s’en détacher et ainsi se trouver sous forme soluble dans le plasma.
 |
Figure 2. Activation de la protéine C. Les accélérations de la réaction d’activation de la protéine C x 20 000 et x 400 000 s’entendent par rapport à la vitesse d’activation de la protéine C par la thrombine (FIIa) en l’absence de cofacteur (TM et/ou EPCR). PC : protéine C ; PCa : PC activée ; TM : thrombomoduline ; EPCR : récepteur endothélial de la protéine C ; PCI : inhibiteur de la protéine C ; 1-AT : 1 antitrypsine ; 2M : 2 macroglobuline. |
Une fois activée, la protéine C (PCa) peut dégrader, par protéolyse limitée, les cofacteurs Va et VIIIa indispensables à la phase d’amplification de la cascade de la coagulation et limiter ainsi la génération de thrombine (Figure 1). Cet effet est potentialisé par la présence d’un cofacteur de la PCa, la protéine S (PS), une autre protéine dont la synthèse requiert la vitamine K [3]. La PS n’accélère la dégradation des FVa et VIIIa par la PCa que si elle est sous forme libre dans le plasma. En effet, près de 60 % de la PS circule liée par son domaine carboxy-terminal à une protéine du système du complément, la C4b binding protein (C4b-BP) [4].
L’importance du système de la PC comme anticoagulant naturel est attestée par la sévérité des signes cliniques développés par les nouveau-nés présentant un déficit homozygote en PC ou en PS. En effet, ces enfants développent un purpura fulminans non infectieux mais comparable à celui qui est observé au cours des méningococcémies de l’enfant. Les déficits hétérozygotes en PC ou PS sont eux associés à la survenue d’évènements thromboemboliques veineux chez l’adulte jeune [5].
L’activité anticoagulante de la PCa est finalement neutralisée dans le plasma par des inhibiteurs de sérine protéase, comme l’inhibiteur de la protéine C, l’alpha 1 anti-trypsine, ou encore l’alpha 2 macroglobuline. La cinétique de cette inhibition est cependant lente puisque, parmi les sérine protéases de la coagulation, la PCa est celle qui a la plus longue demi-vie, environ 15 minutes (Figure 2).
Enfin, en plus de ses propriétés anticoagulantes, la PC possède aussi une activité profibrinolytique, parce qu’elle inhibe le PAI-1 (plasminogen activator inhibitor 1). Comme son nom l’indique, le PAI-1 inhibe l’activation du plasminogène en plasmine, enzyme responsable de la dissolution des caillots formés à l’issue de l’activation de la coagulation [6] (Figure 1).
Physiopathologie du sepsis sévère : une étroite relation entre inflammation et coagulation
Le terme sepsis désigne la réponse systémique de l’organisme à une infection, qui se traduit par les signes d’une réponse inflammatoire systémique (SIRS). Le sepsis est dit sévère lorsqu’existent une hypotension artérielle ou une ou plusieurs défaillances d’organes secondaires à la diminution de la perfusion tissulaire. Lorsque l’hypotension artérielle est réfractaire à l’expansion volémique qui est institutée et nécessite la perfusion d’amines vasopressives, on parle de choc septique. L’incidence et la mortalité du sepsis sont élevées. On estime qu’en Europe 24 % des admissions en réanimation sont liées à un sepsis et que 37,4 % des patients admis en réanimation présentent ou vont présenter un sepsis [7]. Il s’agit d’une complication sévère, dont la mortalité est de l’ordre de 30 % malgré la mise en œuvre des thérapeutiques conventionnelles. En cas de choc septique, la mortalité est supérieure à 50 %.
Sur le plan physiopathologique, c’est la présence de certains composants de la membrane des agents pathogènes, comme le lipopolysaccharide (LPS) de la paroi des bactéries à gram négatif, qui déclenche une réaction inflammatoire explosive et une activation puis un emballement de la coagulation, ces processus étant en relation étroite.
Les modifications induites par l’exposition des monocytes au LPS ont été largement étudiées et impliquent l’activation de la voie de signalisation NF-κB. Cette activation provoque l’augmentation du niveau d’expression de différents gènes dont ceux qui codent pour certaines cytokines pro-inflammatoires (TNF-α [tumor necrosis factor α], interleukine [IL]-1β, IL-6, etc.), pour des molécules d’adhésion (ICAM [intercellular adhesion molecule], VCAM [vascular cell adhesion molecule], etc.) ou encore pour le FT [8] (Figure 1). L’activation du FT déclenche la coagulation et la génération de thrombine. L’activation intense de la coagulation observée au cours du sepsis peut entraîner la consommation de certaines des protéines capables de l’inhiber comme la PC et la PS [9]. Il en résulte un déficit acquis de ces systèmes inhibiteurs de la coagulation qui participe au déséquilibre de la balance hémostatique. Parallèlement, le système fibrinolytique qui devrait permettre de dégrader la fibrine formée au cours de l’activation de la coagulation, se trouve très rapidement inhibé par l’augmentation progressive des taux de PAI-1 dans la circulation. Les défaillances d’organes observées au cours du sepsis sévère sont liées au processus inflammatoire consécutif à l’infection, mais aussi à la formation et au dépôt de fibrine dans la microcirculation, créant des microthrombi. La coagulation tient donc une place essentielle dans le développement de cette pathologie [10].
Les processus inflammatoire et hémostatique impliqués dans le sepsis sévère sont étroitement liés : les médiateurs de l’inflammation tels que le TNF-α, l’IL-6 ou l’IL-1β participeraient à l’activation de la coagulation car ils rendraient l’endothélium vasculaire procoagulant et antifibrinolytique [11]. Ainsi, dans un modèle animal d’endotoxémie, l’inhibition de l’IL-6 réduit considérablement la génération de thrombine [12], alors que l’injection d’IL-1β s’accompagne d’une activation de la coagulation [13]. Par ailleurs, le TNF-α et l’IL-1β modulent la balance fibrinolytique par différents mécanismes dont la stimulation de la production de PAI-1 [13, 14] (Figure 1). L’inhibition de la fibrinolyse qui en résulte contribue à la persistance des dépôts de fibrine et aux défaillances d’organes.
À l’inverse, la coagulation peut fortement moduler l’inflammation. Ainsi, une augmentation de certains médiateurs de l’inflammation pourrait être induite par la thrombine générée au cours de l’activation de la coagulation [15]. En effet la thrombine, mais aussi d’autres sérine protéases de la coagulation comme le FVIIa associé au FT ou le FXa, sont capables d’activer par clivage des récepteurs à 7 domaines transmembranaires de la famille des PAR (protease-activated receptors). Chez l’homme, l’activation du PAR-1 par ces enzymes induit l’activation des monocytes et des cellules endothéliales qui produisent alors des cytokines pro-inflammatoires telles que le TNF-α, l’IL-1β et l’IL-6 (Figure 1). La thrombine est aussi capable de cliver le PAR-1 et le PAR-4 présents à la surface des plaquettes. Ces dernières vont alors exprimer la P-sélectine et ainsi adhérer aux cellules endothéliales et aux leucocytes. L’activation du PAR-2 par le FT/FVIIa induit elle aussi une réponse pro-inflammatoire en activant les macrophages, en favorisant l’infiltration des polynucléaires neutrophiles et l’expression de cytokines pro-inflammatoires [11].
Implication de la protéine C activée dans la physiopathologie du sepsis sévère
Effets cytoprotecteurs de la protéine C
De façon intéressante, la PCa est aussi capable d’activer le PAR-1. C’est par cette voie que la PCa exerce des effets cytoprotecteurs aujourd’hui largement documentés, indépendamment de son rôle crucial dans le maintien de l’équilibre hémostatique (Figure 1). L’implication d’un même récepteur, le PAR-1, dans la transduction d’effets anti-inflammatoires lorsque l’agoniste est la PCa et d’effets pro-inflammatoires lorsque la thrombine est à l’origine de son clivage est surprenante. Ces réponses biologiques opposées pourraient s’expliquer par la capacité qu’a ce récepteur d’activer de façon différentielle plusieurs familles de protéines G et donc de déclencher des voies de signalisation différentes en fonction des agonistes [16].
Ainsi, la PCa réduirait l’inflammation d’une part indirectement en diminuant la génération de thrombine, d’autre part par des mécanismes directs : inhibition de la production de cytokines pro-inflammatoires (IL-6, IL-1β) mais aussi de l’expression de molécules d’adhésion et du FT induites par le TNF-α sur les cellules endothéliales et les leucocytes [17] ; protection de l’intégrité de la barrière endothéliale [18] ; prévention de l’activation des polynucléaires neutrophiles et inhibition de l’adhésion et du rolling des leucocytes [19, 20] (Figure 1). Ces effets passent par une inhibition de la voie NF-κB consécutive au clivage du PAR-1 par la PCa liée soit à l’EPCR exprimé par les cellules endothéliales [21] soit au CD11b présent à la surface des macrophages [22].
Il est maintenant bien établi que l’apoptose est une composante majeure de la physiopathologie du sepsis sévère. Elle touche les lymphocytes, ce qui explique en partie l’immunosuppression, et il semble que les cellules endothéliales soient elles aussi la cible de cet effet pro-apoptotique. Ainsi, l’injection chez la souris de LPS ou de TNF-α stimule l’apoptose des cellules endothéliales [23]. Cela a pour effet de rendre les cellules endothéliales procoagulantes, ce qui pourrait contribuer au dysfonctionnement de l’endothélium et à la constitution des défaillances d’organes [24].
En plus de ses effets anti-inflammatoires, la PCa manifesterait une activité anti-apoptotique. Celle-ci a été mise en évidence d’abord dans un modèle de cellules endothéliales in vitro puis in vivo dans un modèle murin d’ischémie cérébrale. Comme c’était le cas pour ses effets anti-inflammatoires, il semble que la PCa doive se fixer à l’EPCR puis cliver le PAR-1 pour induire un effet anti-apoptotique sur les cellules endothéliales [25]. De même, le clivage du PAR-1 par la PCa, liée cette fois au CD11b à la surface des monocytes [22], est associé à une diminution de l’apoptose de ces cellules, mais aussi à une inhibition de la synthèse de cytokines pro-inflammatoires. Les capacités de phagocytose et d’adhérence, indispensables aux fonctions antimicrobiennes de ces cellules, semblent préservées [26].
Outre la PCa, d’autres composants du système de la PC possèdent une activité anti-inflammatoire. C’est le cas de la TM qui contient un domaine lectine-like aux propriétés anti-inflammatoires. L’implication directe de la TM dans la physiopathologie du sepsis est attestée par des expériences menées chez des souris exprimant une TM dépourvue de domaine lectine (TMLeD/LeD). La TMLeD/LeD est toujours capable d’activer la PC, mais ne peut plus exercer son activité anti-inflammatoire, et les souris exprimant cette TM modifiée ont une sensibilité accrue à l’endotoxine [27]. De la même manière, la PS pourrait moduler la réaction inflammatoire. Du fait de sa forte affinité pour les phospholipides anioniques exposés en particulier par les cellules en apoptose, la PS est capable de se fixer à la surface de ces cellules. Cette liaison aurait deux conséquences : d’une part la stimulation de la phagocytose de ces cellules, évitant ainsi leur rupture et la libération du contenu cellulaire susceptible d’induire une réaction inflammatoire, d’autre part la localisation de la C4b-BP, régulateur de la voie du complément, à la surface des cellules en apoptose [28].
Les histones extracellulaires et le sepsis sévère : une nouvelle cible de la PCa
À l’occasion d’études explorant les effets cytoprotecteurs de la PCa, l’équipe de Charles Esmon [29] a récemment montré que des histones extracellulaires, H3 et H4, sont libérées au cours du sepsis sévère et semblent participer à la physiopathologie de ce dernier [30]. Ainsi, dans un modèle de sepsis chez le primate, l’histone H3 est libérée dans la circulation avec une cinétique comparable à celle de l’augmentation de la créatininémie et de l’installation d’une défaillance rénale. Chez la souris, l’injection de H3, qui possède une cytotoxicité endothéliale in vitro, induit un tableau clinique très proche de celui que crée un sepsis. Dans ce modèle, l’administration de PCa protège les souris de la mortalité induite par l’injection d’une dose létale d’histones. De façon très intéressante, cette équipe a montré que la PCa possède la capacité non seulement de réguler négativement la libération des histones H3 dans la circulation, mais surtout de les cliver aussi bien in vitro qu’in vivo. Contrairement aux autres effets cytoprotecteurs de la PCa, ce clivage ne nécessite pas de liaison de la PCa à l’un de ses récepteurs et ne dépend donc pas de la voie de signalisation de PAR-1.
L’implication de ces histones extracellulaires dans la physiopathologie du sepsis sévère chez l’homme reste cependant à démontrer, même si des histones H3 sont détectées dans le plasma de patients lors de sepsis sévère [29].
Protéine C activée et traitement du sepsis sévère
Comme nous l’avons vu, la PCa possède de nombreux atouts (activités anticoagulante, anti-inflammatoire, anti-apoptotique, et anti-histone) pour constituer un traitement de choix du sepsis sévère. De plus, au cours du sepsis sévère, la majorité des patients présentent des taux diminués de PC, ce qui est un critère de mauvais pronostic [31]. Cet argument, associé aux multiples propriétés de la PCa, ont justifié l’utilisation de concentrés de PC plasmatique purifiée dans le traitement du sepsis. Ainsi, chez les babouins, l’administration de PC était capable de réduire la mortalité des animaux chez lesquels on avait induit un sepsis à Escherichia coli [32]. Théoriquement, l’avantage de ce procédé était qu’il permettait d’activer le système anticoagulant (la PC exogène) proportionnellement à la quantité de thrombine formée, et donc de contrôler finement le processus. Cependant, l’altération de l’endothélium suspectée dans le sepsis sévère faisant craindre une incapacité de certains patients à activer cette PC exogène, la PCa s’est progressivement substituée à la PC dans les essais cliniques.
L’ensemble de ces observations a donc justifié la mise en place d’essais thérapeutiques chez l’homme basés sur l’utilisation de la PCa dans le sepsis. Les principaux résultats des études cliniques sur lesquels sont fondées les recommandations de l’utilisation de la PCa dans le traitement du sepsis sévère sont détaillés dans l’Encadré.
Cependant, une des limitations de cette approche est liée à l’activité anticoagulante de la PCa et donc au risque hémorragique associé à l’utilisation de cette protéine. La connaissance de plus en plus fine de la relation structure-fonction de la PC a permis à une équipe de concevoir des variants de la PCa toujours doués de propriétés cytoprotectrices, mais dont l’activité anticoagulante est réduite [33]. Ces variants, en cours d’étude, ouvrent de nouvelles perspectives dans l’utilisation de cette thérapeutique innovante.
Les essais cliniques de la PCa dans le sepsis
PCa : un médicament très attendu
En 2001, l’étude PROWESS publiée dans le New England Journal of Medicine décrit l’efficacité de l’administration d’une PCa recombinante dans la prise en charge du sepsis sévère et du choc septique [34]. Il s’agissait enfin de la première étude positive permettant de proposer un traitement spécifique du sepsis, en dehors des antibiotiques. L’étude comprenait plus de 1 600 patients : les investigateurs y comparaient un groupe contrôle à un groupe de patients bénéficiant d’une perfusion de PCa recombinante débutée dans les 48 heures suivant le début du sepsis [34]. L’administration était réalisée par voie intraveineuse continue à la seringue électrique, à la dose de 24 µg/kg/heure, pour une durée de perfusion de 96 heures. La mortalité au jour 28 des patients du groupe traité était significativement diminuée, passant de 30,8 % à 24,7 %, pour une réduction relative du risque de décès de 19,4 % [6,6 - 30,5, 95 % intervalle de confiance] [34]. L’effet était d’autant plus marqué que l’état des patients nécessitait une ventilation mécanique, la perfusion d’amines vasopressives et que le nombre de défaillances d’organes était important [35]. À la suite de cette étude, une autorisation de mise sur le marché était délivrée pour la PCa recombinante aux États-Unis et en Europe. Les indications étaient un peu différentes selon le continent : aux États-Unis, le médicament était recommandé chez les patients septiques ayant un score de gravité APACHE II > 25 (acute physiology and chronic health evaluation, score d’évaluation des patients en réanimation) ; en Europe, chez les patients septiques ayant au moins 2 défaillances d’organes.
Effets secondaires et précautions d’emploi
La perfusion de PCa a des effets secondaires, notamment hémorragiques, du fait de son activité anticoagulante et profibrinolytique. Dans l’étude PROWESS, 2 % des patients du groupe contrôle présentaient un événement hémorragique sévère durant les 28 premiers jours contre 3,5 % dans le groupe traité avec la PCa (p = 0,06) [34]. Dans une autre étude ouverte (ENHANCE), simple bras, portant sur 2 378 patients ayant reçu de la PCa recombinante, cette proportion était de 6,5 % [36]. Tant dans PROWESS que dans ENHANCE, l’incidence des saignements intracrâniens, ceux dont le pronostic est le plus sombre, était faible, 0,2 % et 1,5 % respectivement. Ces effets secondaires hémorragiques ont conduit les autorités à contre-indiquer la perfusion de PCa recombinante chez les patients particulièrement à risque de saignement, ou porteurs d’une pathologie intracrânienne, ou ceux dont le taux de plaquettes était inférieur à 30 000/mm3. En pratique, un patient traité par la PCa recombinante et dont le taux de plaquettes décroît en dessous de 30 000/mm3 doit bénéficier d’une transfusion de plaquettes. Dans la pratique courante, toute procédure invasive, comme la mise en place d’un cathéter veineux central ou d’un drain thoracique, doit s’accompagner d’une interruption de la perfusion de PCa 2 heures avant la procédure et se prolongeant jusqu’à 6 heures après. Il semble cependant qu’une reprise immédiate du produit soit possible mais des résultats permettant de le confirmer sont en attente. Un sepsis ayant un point de départ abdominal devra faire l’objet, avant la prescription de PCa recombinante, d’un traitement chirurgical adapté avec une hémostase minutieuse, moyennant quoi le risque hémorragique ne semble pas augmenté dans ce sous-groupe de patients [37]. Un délai de 12 heures entre le geste chirurgical et la prescription du produit est néanmoins recommandé. Enfin, une héparinothérapie [42] concomitante est possible, mais seulement à dose préventive (<10 000 unités/24 heures). À ce propos, il semble recommandé de ne pas arrêter complètement une héparinothérapie qui aurait été instaurée préalablement à la prescription de PCa. Cela exposerait en effet à une augmentation des événements ischémiques [5].
Le temps de la controverse
Il faut cependant souligner quelques points-clés et sujets de controverse concernant la prescription de PCa recombinante. Le plus important, et il a passionné la communauté scientifique, porte sur l’efficacité réelle ou supposée du produit. Pour lever cette incertitude, les autorités ont demandé des études complémentaires pour confirmer, ou infirmer, les résultats positifs de l’étude PROWESS. Deux essais randomisés contre placebo sont en cours d’inclusion, un institutionnel et l’autre émanant du laboratoire pharmaceutique ayant commercialisé le médicament, appelé PROWESS SHOCK [38]. Dans les 2 cas, les patients inclus doivent présenter un choc septique et non un sepsis sévère, leur mortalité attendue étant ainsi plus élevée que dans l’étude PROWESS.
Le deuxième point de débat concerne en effet la sélection des patients. Une étude plus récente, ADDRESS, a rapporté que la perfusion de PCa recombinante à des patients septiques ayant un « faible » risque de mortalité n’entraînait aucun bénéfice en terme de survie [39]. À l’usage, l’indication du produit basée sur un score APACHE II > 25 ou sur la présence d’au moins 2 défaillances d’organes est apparue trop large. À partir des données des différentes études cliniques, il s’avère que l’effet du produit sur la mortalité était surtout présent chez les patients ayant 3, 4 (ou plus) défaillances d’organe [40]. Les investigateurs de l’étude PROWESS ont proposé un score permettant de calculer un risque de mortalité, basé sur l’âge, le score APACHE II, la présence de comorbidité, le taux d’IL-6 et l’origine du sepsis [35]. Ils ont démontré que l’effet de la PCa recombinante était significatif lorsque le risque de mortalité devenait supérieur à 40 %, l’effet étant maximal au-delà de 60 % de mortalité attendue [35]. En pratique, ce score est cependant difficile à utiliser. Nous réservons le médicament aux patients intubés ventilés, nécessitant des doses croissantes d’amines vasopressives et chez lesquels persiste une acidose lactique malgré l’optimisation de la prise en charge [41]. Chez ces patients particulièrement graves, la PCa recombinante semble permettre une baisse de mortalité de plus de 50 % [41], même si ces résultats doivent être analysés à la lumière des limites méthodologiques de l’étude qui est une étude rétrospective avant/après.
Un autre point-clé concerne le moment de la perfusion. Les données de la littérature insistent sur la nécessité de sélectionner les patients dans les 24 heures suivant le début du sepsis. Vincent et al. ont montré que l’effet s’estompait au-delà de la 24e heure [7] pour probablement devenir négligeable au-delà d’un certain délai. En pratique, les 12 premières heures représentent toujours le temps des prélèvements bactériologiques, de l’administration des antibiotiques, du traitement chirurgical si besoin, de l’optimisation hémodynamique (cathéters veineux central et artériel, remplissage, amines vasopressives) et respiratoire (intubation, ventilation mécanique). Au décours de ces 12 premières heures, l’évaluation de la gravité et de la réponse au traitement optimisé permet la prescription du produit aux bons patients dans le bon timing [41].
Enfin, compte tenu de ses effets secondaires pro-hémorragiques, une évaluation de la balance bénéfice/risque de la PCa recombinante doit être réalisée régulièrement. Même si la durée du traitement recommandée est de 96 heures, il n’est pas rare de décider l’arrêt de la perfusion après 72 heures par exemple chez un patient sevré d’amines vasopressives, bien que les données de la littérature soient parcellaires sur ce point.
En conclusion, après l’enthousiasme initial suscité par la publication de l’étude PROWESS, beaucoup de questions ont été soulevées concernant d’une part l’efficacité réelle de la PCa et d’autre part les conditions de son utilisation chez les patients septiques. Dans l’attente des résultats des deux grands essais en cours, il est cependant suggéré dans la littérature : (1) de proposer ce médicament aux patients les plus graves, en choc septique, plutôt que dans le sepsis sévère ; (2) que le moment de sa prescription est fondamental (ni trop tôt, pour correctement sélectionner les patients, ni trop tard chez des patients ayant un état dépassé) ; et (3) que l’évaluation de la balance bénéfice/risque est également cruciale et doit être répétée afin d’interrompre un traitement devenu inutile ou dangereux.
Conflit d’intérêts
D. Borgel déclare n’avoir aucun conflit d’intérêts concernant les données publiées dans cet article.
A. Veillard-Baron déclare avoir participé à des conférences, colloques et actions de formation pour l’entreprise Lilly.
Références
- Esmon CT. The roles of protein C and thrombomodulin in the regulation of blood coagulation. J Biol Chem 1989 ; 264 : 4743-4746. [PubMed] [Google Scholar]
- Gandrille S, Saposnik B, Aiach M. Le récepteur endothélial à la protéine C (EPCR) : un récepteur à l’interface entre coagulation et système inflammatoire. Hématologie 2001 ; 7 : 418-28. [Google Scholar]
- Esmon CT, Fukudome K. Cellular regulation of the protein C pathway. Semin Cell Biol 1995 ; 6 : 259-268. [CrossRef] [PubMed] [Google Scholar]
- Dahlback B. Protein S and C4b-binding protein : components involved in the regulation of the protein C anticoagulant system. Thromb Haemost 1991 ; 66 : 49-61. [PubMed] [Google Scholar]
- Aiach M, Alhenc-Gelas M, Borgel D, et al. Mutations des protéines de la coagulation et thromboses. Med Sci (Paris) 2006 ; 22 : 985-989. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Rezaie AR. Vitronectin functions as a cofactor for rapid inhibition of activated protein C by plasminogen activator inhibitor-1. Implications for the mechanism of profibrinolytic action of activated protein C. J Biol Chem 2001 ; 276 : 15567-15570. [CrossRef] [PubMed] [Google Scholar]
- Vincent JL, Sakr Y, Sprung CL, et al. Sepsis in European intensive care units : results of the SOAP study. Crit Care Med 2006 ; 34 : 344-353. [CrossRef] [PubMed] [Google Scholar]
- Guha M, Mackman N. LPS induction of gene expression in human monocytes. Cell Signal 2001 ; 13 : 85-94. [CrossRef] [PubMed] [Google Scholar]
- Levi M, de Jonge E, van der Poll T. Rationale for restoration of physiological anticoagulant pathways in patients with sepsis and disseminated intravascular coagulation. Crit Care Med 2001 ; 29 : S90-S94. [CrossRef] [PubMed] [Google Scholar]
- Gando S. Microvascular thrombosis and multiple organ dysfunction syndrome. Crit Care Med 2010 ; 38 : S35-S42. [CrossRef] [PubMed] [Google Scholar]
- Levi M, van der Poll T. Two-way interactions between inflammation and coagulation. Trends Cardiovasc Med 2005 ; 15 : 254-259. [CrossRef] [PubMed] [Google Scholar]
- van der Poll T, Levi M, Hack CE, et al. Elimination of interleukin 6 attenuates coagulation activation in experimental endotoxemia in chimpanzees. J Exp Med 1994 ; 179 : 1253-1259. [CrossRef] [PubMed] [Google Scholar]
- Jansen PM, Boermeester MA, Fischer E, et al. Contribution of interleukin-1 to activation of coagulation and fibrinolysis, neutrophil degranulation, and the release of secretory-type phospholipase A2 in sepsis : studies in nonhuman primates after interleukin-1 alpha administration and during lethal bacteremia. Blood 1995 ; 86 : 1027-1034 [PubMed] [Google Scholar]
- Sawdey MS, Loskutoff DJ. Regulation of murine type 1 plasminogen activator inhibitor gene expression in vivo. Tissue specificity and induction by lipopolysaccharide, tumor necrosis factor-alpha, and transforming growth factor-beta. J Clin Invest 1991 ; 88 : 1346-1353. [CrossRef] [PubMed] [Google Scholar]
- Esmon CT. Inflammation and thrombosis. J Thromb Haemost 2003 ; 1 : 1343-1348. [CrossRef] [PubMed] [Google Scholar]
- McLaughlin JN, Shen L, Holinstat M, et al. Functional selectivity of G protein signaling by agonist peptides and thrombin for the protease-activated receptor-1. J Biol Chem 2005 ; 280 : 25048-25059. [CrossRef] [PubMed] [Google Scholar]
- Joyce DE, Nelson DR, Grinnell BW. Leukocyte and endothelial cell interactions in sepsis : relevance of the protein C pathway. Crit Care Med 2004 ; 32 : S280-S286. [CrossRef] [PubMed] [Google Scholar]
- Feistritzer C, Riewald M. Endothelial barrier protection by activated protein C through PAR1-dependent sphingosine 1-phosphate receptor-1 crossactivation. Blood 2005 ; 105 : 3178-3184. [CrossRef] [PubMed] [Google Scholar]
- Sturn DH, Kaneider NC, Feistritzer C, et al. Expression and function of the endothelial protein C receptor in human neutrophils. Blood 2003 ; 102 : 1499-1505. [CrossRef] [PubMed] [Google Scholar]
- Elphick GF, Sarangi PP, Hyun YM, et al. Recombinant human activated protein C inhibits integrin-mediated neutrophil migration. Blood 2009 ; 113 : 4078-4085. [CrossRef] [PubMed] [Google Scholar]
- Riewald M, Petrovan RJ, Donner A, et al. Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science 2002 ; 296 : 1880-1882. [CrossRef] [PubMed] [Google Scholar]
- Cao C, Gao Y, Li Y, et al. The efficacy of activated protein C in murine endotoxemia is dependent on integrin CD11b. J Clin Invest 2010 ; 120 : 1971-1980. [CrossRef] [PubMed] [Google Scholar]
- Mahidhara R, Billiar TR. Apoptosis in sepsis. Crit Care Med 2000 ; 28 : N105-N113. [CrossRef] [PubMed] [Google Scholar]
- Aird WC. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood 2003 ; 101 : 3765-3777. [CrossRef] [PubMed] [Google Scholar]
- Cheng T, Liu D, Griffin JH, et al. Activated protein C blocks p53-mediated apoptosis in ischemic human brain endothelium and is neuroprotective. Nat Med 2003 ; 9 : 338-342. [CrossRef] [PubMed] [Google Scholar]
- Stephenson DA, Toltl LJ, Beaudin S, Liaw PC. Modulation of monocyte function by activated protein C, a natural anticoagulant. J Immunol 2006 ; 177 : 2115-2122. [PubMed] [Google Scholar]
- Conway EM, Van de Wouwer M, Pollefeyt S, et al. The lectin-like domain of thrombomodulin confers protection from neutrophil-mediated tissue damage by suppressing adhesion molecule expression via nuclear factor kappaB and mitogen-activated protein kinase pathways. J Exp Med 2002 ; 196 : 565-577. [CrossRef] [PubMed] [Google Scholar]
- Anderson HA, Maylock CA, Williams JA, et al. Serum-derived protein S binds to phosphatidylserine and stimulates the phagocytosis of apoptotic cells. Nat Immunol 2003 ; 4 : 87-91. [CrossRef] [PubMed] [Google Scholar]
- Xu J, Zhang X, Pelayo R, et al. Extracellular histones are major mediators of death in sepsis. Nat Med 2009 ; 15 : 1318-1321. [CrossRef] [PubMed] [Google Scholar]
- Viatte L Dr Jekyll et Mr Histone : protéger ou tuer, la dualité des histones. Med Sci (Paris) 2010 ; 26 : 144. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Shorr AF, Bernard GR, Dhainaut JF, et al. C Protein concentrations in severe sepsis : an early directional change in plasma levels predicts outcome. Crit Care 2006 ; 10 : R92. [CrossRef] [PubMed] [Google Scholar]
- Taylor FB, Jr., Chang A, Esmon CT, et al. Protein C prevents the coagulopathic and lethal effects of Escherichia coli infusion in the baboon. J Clin Invest 1987 ; 79 : 918-925. [CrossRef] [PubMed] [Google Scholar]
- Kerschen EJ, Fernandez JA, Cooley BC, et al. Endotoxemia and sepsis mortality reduction by non-anticoagulant activated protein C. J Exp Med 2007 ; 204 : 2439-2448. [CrossRef] [PubMed] [Google Scholar]
- Bernard GR, Vincent JL, Laterre PF, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med 2001 ; 344 : 699-709. [CrossRef] [PubMed] [Google Scholar]
- Ely EW, Laterre PF, Angus DC, et al. Drotrecogin alfa (activated) administration across clinically important subgroups of patients with severe sepsis. Crit Care Med 2003 ; 31 : 12-19. [CrossRef] [PubMed] [Google Scholar]
- Vincent JL, Bernard GR, Beale R, et al. Drotrecogin alfa (activated) treatment in severe sepsis from the global open-label trial ENHANCE : further evidence for survival and safety and implications for early treatment. Crit Care Med 2005 ; 33 : 2266-2277. [CrossRef] [PubMed] [Google Scholar]
- Payen D, Sundin DP, Nelson DR, Williams MD. Analysis of efficacy and safety of drotrecogin alfa (activated) in surgical patients, using an international integrated database. Surgery 2007 ; 142 : 426-427. [CrossRef] [Google Scholar]
- Thompson BT, Ranieri VM, Finfer S, et al. Statistical analysis plan of PROWESS SHOCK study. Intensive Care Med 2010 ; 36 : 1972-1973. [CrossRef] [PubMed] [Google Scholar]
- Abraham E, Laterre PF, Garg R, et al. Drotrecogin alfa (activated) for adults with severe sepsis and a low risk of death. N Engl J Med 2005 ; 353 : 1332-1341. [CrossRef] [PubMed] [Google Scholar]
- Carlet J. Prescribing indications based on successful clinical trials in sepsis : a difficult exercise. Crit Care Med 2006 ; 34 : 525-529. [CrossRef] [PubMed] [Google Scholar]
- Vieillard-Baron A, Caille V, Charron C, et al. Reversal of refractory septic shock with drotrecogin alpha (activated). Intensive Care Med 2009 ; 35 : 1204-1209. [CrossRef] [PubMed] [Google Scholar]
- Dumont B, Faille D, Ajzenberg N. Les nouveaux anticoagulants oraux : dabigatran, rivaroxaban et apixaban. Leur utilisation actuelle et leur avenir. Med Sci (Paris) 2011 ; 27 : 493-500. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
Liste des figures
|
Figure 1. Effets de la protéine C activée au cours du sepsis. Les traits se terminant par une flèche correspondent à un effet activateur, les traits se terminant par un rond à un effet inhibiteur. Les traits rouges correspondent aux effets de la protéine C activée. PC : protéine C ; PCa : PC activée ; FT : facteur tissulaire ; TM : thrombomoduline ; EPCR : récepteur endothélial de la protéine C ; tPA : activateur tissulaire du plasminogène ; PAI-1 : inhibiteur de l’activateur du plasminogène ; H3 : histone extracellulaire 3 ; VCAM : vascular cell adhesion molecule ; ICAM : intercellular cell adhesion molecule ; IL-6, IL-1β : interleukines ; TNF-α : tumor necrosis factor. |
| Dans le texte | |
|
Figure 2. Activation de la protéine C. Les accélérations de la réaction d’activation de la protéine C x 20 000 et x 400 000 s’entendent par rapport à la vitesse d’activation de la protéine C par la thrombine (FIIa) en l’absence de cofacteur (TM et/ou EPCR). PC : protéine C ; PCa : PC activée ; TM : thrombomoduline ; EPCR : récepteur endothélial de la protéine C ; PCI : inhibiteur de la protéine C ; 1-AT : 1 antitrypsine ; 2M : 2 macroglobuline. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.