")
")
| Issue |
Med Sci (Paris)
Volume 26, Number 12, Décembre 2010
|
|
|---|---|---|
| Page(s) | 1061 - 1066 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/201026121061 | |
| Published online | 15 décembre 2010 | |
Cellules souches embryonnaires humaines et iPS
Une source fiable d’hépatocytes fœtaux
Robust differentiation of fetal hepatocytes from human embryonic stem cells and iPS
1
Inserm U972, IFR 93, Hôpital du Kremlin-Bicêtre, 78, rue du Général Leclerc, 94270 Le Kremlin-Bicêtre, France
2
Université Paris-Sud, F-91405 Orsay, France
3
Department of Surgery, The Anne McLaren Laboratory for Regenerative Medicine, Cambridge, CB2 0SZ, Royaume-Uni
Résumé
La transplantation d’hépatocytes est considérée comme une alternative à la transplantation d’organes pour le traitement de maladies métaboliques notamment. Cependant, en raison des difficultés de disposer d’un grand nombre d’hépatocytes, de nouvelles sources de cellules ont été envisagées. Ces cellules peuvent être d’origine hépatique (cellules souches hépatiques) ou extra-hépatique, telles les cellules souches mésenchymateuses ou les cellules souches pluripotentes (cellules souches embryonnaires humaines [CSEh] ou iPS). Nous avons mis au point une méthode de différenciation des CSEh en hépatocytes fœtaux. Les conditions de différenciation qui reproduisent les étapes majeures du développement embryonnaire du foie sont établies en milieu défini, sans ajout de produit d’origine animale ou indéterminée. Les cellules obtenues expriment de nombreux marqueurs d’hépatocytes fœtaux (cytochrome p450 3A7, albumine, alpha-1-antitrypsine, etc.). Elles sont capables d’assurer des fonctions spécifiques des hépatocytes (métaboliser l’ammonium, excréter le vert d’indocyanine), et enfin de se développer et d’exprimer des protéines hépatiques deux mois après transplantation dans le foie de souris nouveau-nés immunodéficientes (uPAxrag2γc−/−). Nous avons également démontré que ce protocole s’appliquait également aux iPS, et les études dans les modèles animaux nous permettront de comparer le potentiel in vivo de ces deux sources de cellules pluripotentes. Seules des approches précliniques effectuées chez le primate permettront de valider une éventuelle application à l’homme.
Abstract
Hepatocyte transplantation is considered as an alternative to organ transplantation in particular for the treatment of liver metabolic diseases. However, due to the difficulties to obtain a large number of hepatocytes, new sources of cells are needed. These cells could be either of hepatic origin (hepatic stem cells) or extrahepatic such as mesenchymal stem cells or pluripotent stem cells (human embryonic stem cells [hESC] or iPS). We developed a new method to differentiate hESCs into fetal hepatocytes. These conditions recapitulate the main liver developmental stages, using fully defined medium devoid of animal products or unknown factors. The differentiated cells express many fetal hepatocytes markers (cytochrome P450 3A7, albumin, alpha-1-antitrypsin, etc.). The cells display specific hepatic functions (ammonia metabolism, excretion of indocyanin green) and are capable to engraft and express hepatic proteins two months after transplantation into newborn uPA x rag2γc−/− mouse liver. We have also showed that this approach is transposable to human iPS, and further studies on animal models will allow us to compare the in vivo potential of these two sources of pluripotent cells. Finally, only studies on large animals such as nonhuman primates will validate an eventual clinical application.
© 2010 médecine/sciences - Inserm / SRMS
Le foie est le siège de nombreux déficits métaboliques (maladies du cycle de l’urée, hypercholestérolémie familiale, syndrome de Crigler-Najjar type I, etc.), mais aussi la cible d’atteintes hépatiques chroniques ou aiguës. Actuellement la transplantation orthotopique est le seul traitement curatif des maladies métaboliques sévères engageant le pronostic vital mais elle est restreinte par la pénurie croissante de donneurs d’organe. La thérapie cellulaire apparaît depuis quelques années comme une alternative thérapeutique pour le traitement de ces maladies. En effet, elle permettrait de remplacer les seuls hépatocytes porteurs d’un défaut génétique, en laissant intact le foie. L’obstacle majeur de cette approche est que les hépatocytes ne peuvent pas être amplifiés en culture et résistent mal à la congélation. Il est donc nécessaire d’évaluer le potentiel d’autres sources de cellules, qui non seulement pourraient se différencier en hépatocytes mais aussi proliférer in vitro. Ces deux propriétés caractérisent les cellules souches pluripotentes, et en particulier les cellules souches embryonnaires et les iPS (induced pluripotent stem cells). Nous faisons le point dans cette revue sur les progrès réalisés dans l’obtention d’hépatocytes fonctionnels à partir de cellules souches pluripotentes en nous fondant sur l’expérience de notre équipe dans ce domaine.
Différenciation hépatique des cellules ES
Après les premières tentatives de différenciation via la formation de corps embryoïdes, les stratégies ont consisté à différencier les CSEh séquentiellement, en suivant trois étapes physiologiques résumées sur la Figure 1.
Spécification des CSEh en cellules de l’endoderme définitif. Ce stade préalable à leur orientation vers un devenir hépatique est induit par l’activine, dont l’importance de la voie de signalisation a été mise en évidence en utilisant des cellules murines et humaines : l’équipe de K.A. d’Amour a montré la première que l’exposition des CSEh à une forte concentration d’activine pendant 3 jours puis à Wnt3a pendant 24 heures en présence de sérum induisait la différenciation endodermique de 80 % des cellules [1].
Spécification hépatique. Elle est contrôlée par l’ajout de molécules destinées à rendre les progéniteurs endodermiques sensibles aux signaux inducteurs de la différenciation hépatique. Ces molécules peuvent être des cytokines, telles Wnt3a [2], FGF4/BMP2 (fibroblast growth factor/bone morphogenic protein) [3] ou BMP4/FGF2, et elles sont généralement couplées soit à la culture en conditions hypoxiques [4] soit au sodium butyrate, inhibiteur de l’acétylation des histones [5], ou au DMSO (diméthylsulfoxide) induisant la méthylation de l’ADN [6].
Différenciation hépatique. Elle est induite par l’ajout de HGF (hepatocyte growth factor) puis de dexaméthasone et d’oncostatine. Les cellules obtenues expriment de nombreux marqueurs hépatiques, par exemple CK8/18, AFP (α-fœtoprotéine), AAT (α1-antitrypsine), HNF4α (hepatic nuclear factor) et montrent une certaine fonctionnalité in vitro comme le stockage du glycogène ou une faible activité de certains cytochromes P450 [2]. Toutefois, dans les rares cas où la fonctionnalité des cellules a été analysée in vivo, elle l’a été à très court terme, 5 jours après l’injection des cellules ; à plus long terme, les cellules ont entraîné la formation de tératomes voire d’adénocarcinomes [2, 4, 6], probablement en raison de la persistance de CSEh indifférenciées parmi les cellules greffées.
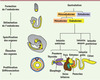 |
Figure 1 Origine et formation de l’endoderme. À gauche : résumé des événements se produisant lors de la formation de l’endoderme qui s’accompagne d’un changement de morphologie de l’embryon de souris. L’endoderme est représenté en jaune. L’endoderme et le mésoderme dériveraient d’un progéniteur commun, le mésendoderme. À droite : différents lignages dérivés de l’endoderme projetés sur une représentation du tractus gastrointestinal (d’après [46]). |
Un protocole de différenciation hépatique des CSEh compatible avec une application clinique
Les approches résumées ci-dessus sont toutes basées sur des conditions de culture utilisant du sérum, des matrices complexes (matrigel), des sous-couches de cellules nourricières, voire des agents affectant de façon aspécifique le profil épigénétique des cellules. Elles mettent de ce fait en jeu des facteurs inconnus qui interfèrent avec les mécanismes moléculaires contrôlant le développement du foie et qui sont incompatibles avec une utilisation clinique ultérieure.
Des conditions définies mimant le développement de l’ébauche hépatique embryonnaire
Afin de répondre à ces contraintes, nous avons récemment publié une nouvelle approche pour induire la différenciation des CSEh en hépatocytes. Celle-ci utilise des conditions chimiquement définies et suit les étapes clefs du développement hépatique [7] (Figures 1 et 2).
Les trois étapes sont identiques à celles précédemment définies.
Induction des CSEh pluripotentes en cellules de l’endoderme définitif. Cette étape est contrôlée par une combinaison de 4 facteurs, nécessaires mais suffisants, utilisée pendant 3 jours : concentration élevée d’activine A, de BMP4 et de FGF2, facteurs connus pour contrôler la spécification des 3 lignées germinales chez les mammifères, auxquels est ajouté du LY294004 (un inhibiteur chimique de la PI3K) [8]. Le milieu utilisé est un milieu défini (AFBLy) [9] dépourvu de sérum mais contenant différents suppléments dont l’insuline et l’alcool polyvinylique, substitut de l’albumine sérique. Dans ces conditions, non seulement les marqueurs de pluripotence étaient réprimés mais 80 % des cellules exprimaient fortement les marqueurs de l’endoderme (Sox17, GSC [goosecoid], Mixl1 [Mix1 homeobox-like 1], Lhx1 [LIM homeobox protein 1], CXCR4 [récepteur de chimiokine], GATA6). De plus l’absence de Sox7, marqueur du tissu extra-embryonnaire, et de Sox2, marqueur du neurectoderme, exclut que nos conditions aient induit des cellules de l’endoderme primitif ou des progéniteurs neuronaux.
Spécification hépatique. Les cellules de l’endoderme définitif ont été cultivées 3 jours en présence de FGF10, puis 2 jours en présence de FGF10 et d’acide rétinoïque, ces deux facteurs étant connus pour leur rôle critique dans la croissance du foie et la survie des hépatocytes in vivo [10, 11]. L’ajout de SB431542, un inhibiteur pharmacologique des récepteurs à l’activine/Nodal, a permis d’augmenter l’expression des marqueurs de spécification. Les cellules de l’endoderme définitif exprimaient un niveau élevé de Wnta, confirmant des résultats déjà publiés sur le rôle essentiel de Wnta dans les mécanismes contrôlant la spécification des cellules d’endoderme définitif en endoderme hépatique.
Obtention d’hépatoblastes. L’ajout de plusieurs facteurs (FGF4, HGF et EGF) pendant 5 jours a induit la prolifération d’une population de cellules exprimant des marqueurs de progéniteurs hépatiques : environ 50 % des cellules co-exprimaient l’AFT et la cytokératine 19 (CK19), marqueur des hépatocytes et des cellules biliaires respectivement, suggérant que cette population correspond aux hépatoblastes, progéniteurs communs à ces deux types cellulaires dans le foie fœtal. Les cellules exprimaient également EpCam (epithelial cell adhesion molecule), un marqueur de cellules souches/progénitrices du foie fœtal [12, 43] et des facteurs de transcription hépatique tel HNF4a - connu pour contrôler l’expression de nombreux gènes spécifiques [13] (Figure 2). Ces cellules ont été transplantées dans des souris immunodéficientes1 uPAxRagγc−/− transgéniques pour l’urokinase (aussi appelée activateur du plasminogène ou PA). Chez ces animaux, l’expression de l’urokinase est toxique pour les hépatocytes endogènes, ce qui bloque transitoirement la régénération et permet une meilleure prise de greffe des cellules transplantées. Huit semaines après transplantation, des ilôts de cellules co-exprimant l’albumine ou l’alpha-1-antitrypsine humaine et la GFP (green fluorescent protein) ont été mis en évidence sans apparition concomitante de tératomes. Ainsi, les cellules hépatiques dérivées des CSEh sont non seulement capables de greffer chez l’hôte receveur et de rester fonctionnelles, mais aussi de proliférer in situ dans ce foie receveur.
La différenciation des progéniteurs en hépatocytes. Elle a été induite in vitro par l’ajout d’hydrocortisone, d’une combinaison de facteurs de croissance (FGF4, EGF et HGF), connus pour être impliqués dans la croissance du foie fœtal, et d’oncostatine. Après huit jours de culture dans ces conditions, les cellules hépatiques exprimaient plusieurs protéines caractéristiques des fonctions hépatocytaires comme l’α1-anti-trypsine, le récepteur des low density lipoproteins, l’apolipoprotéine A-II (ApoAII), la tyrosine aminotransférase, la tryptophane 2,3-dioxygénase, le récepteur aux asialoglycoprotéines de type 1, le facteur IX de coagulation et les cytochromes P450, CYP3A7 et Cyp7A1 spécifiques du foie fœtal. Tous ces marqueurs étaient exprimés de façon similaire à leur expression dans des cellules hépatiques fœtales primaires (Figure 2).
 |
Figure 2 Différenciation des CSEh en hépatocytes. Les CSEh indifférenciées progressent dans la voie hépatique par étapes successives correspondant à l’expression transitoire de certains marqueurs comme Sox17 et CXCR4 au stade de l’endoderme définitif (DE), EpCam et N-Cam au stade des cellules souches hépatiques (hHpSC). Au cours de leur différenciation, les cellules changent de morphologie pour adopter, au stade hépatocytes, une forme pentagonale caracté- ristique. FC : fetal cholangiocytes ; FH : fetal hepatocytes. |
Analyse de la fonction des hépatocytes obtenus
En termes de fonctionnalité in vitro, les hépatocytes dérivés des CSEh étaient capables de sécréter de l’urée et de l’albumine, d’internaliser puis d’excréter le vert d’indocyanine (fonctions spécifiques des hépatocytes) et montraient une activité CYP3A7. L’ensemble de ces résultats, incluant l’absence du CYP3A4, suggère que les cellules ainsi différenciées correspondent à des hépatocytes fœtaux. L’absence dans nos expériences de formation de tératomes ou d’adénocarcinomes alors que nous n’avons pas inclus d’étape finale de purification suggère que nos conditions induisent bien l’engagement dans la voie de différenciation hépatocytaire de la totalité des cellules pluripotentes.
Étape ultime de différenciation en hépatocytes adultes matures
La maturation d’hépatocytes fœtaux en hépatocytes adultes nécessitera probablement la mise en place de cocultures, les hépatocytes fonctionnant in vivo en étroites interactions avec les autres cellules du foie. Si la génération d’hépatocytes complètement différenciés (qui expriment les enzymes de détoxification de phases I et II) est nécessaire pour les études de pharmacotoxicologie ou pour la mise au point de foies bioartificiels, ce n’est en revanche pas le cas pour les approches de thérapie cellulaire des maladies métaboliques hépatiques. En effet, de nombreuses études chez les rongeurs et le primate ont montré une faible efficacité (< 50 %) de la prise de greffe d’hépatocytes adultes et l’absence de prolifération de ces cellules in situ. L’utilisation de progéniteurs de type hépatoblastes pourrait être une alternative plus efficace. La preuve de concept a été établie avec les précurseurs neuronaux [14] et nous avons montré que des progéniteurs fœtaux pouvaient proliférer et se différencier dans le foie de souris [15]. De nombreux modèles murins sont encore nécessaires pour mieux caractériser le devenir des cellules dérivées des CSEh in vivo après transplantation. Mais en aucun cas ils ne pourront être suffisants, et des approches précliniques sur le primate devront être développées avant d’envisager toute application thérapeutique. Après le report initial des essais prévus par Geron, le premier patient atteint d’une lésion de la moelle épinière vient d’être traité aux États-Unis en octobre 2010 par des progéniteurs d’oligodendrocytes dérivés de cellules souches embryonnaires humaines. D’autres essais cliniques de phases I/II sont prévus aux États-Unis et en Angleterre en 2011 pour le traitement de patients atteints de dégénérescence maculaire avec les cellules hES différenciées [16]. Notons qu’en France, l’équipe de Michel Pucéat et Philippe Ménasché progresse dans l’évaluation d’un protocole thérapeutique utilisant des progéniteurs cardiaques dérivés de CSEh récemment évalués dans le modèle préclinique du macaque [44]. Le long cheminement nécessaire pour aboutir à des applications cliniques de ces cellules est donc désormais amorcé aussi en France.
Les cellules pluripotentes induites (iPS) peuvent-elles remplacer les cellules ES ?
La reprogrammation de cellules somatiques humaines et murines comme les fibroblastes [17, 18, 45] mais aussi les lymphocytes B matures, les cellules épithéliales de l’estomac et les hépatocytes [19] a ouvert la voie à la génération de cellules pluripotentes reprogrammées spécifiques de patients. Outre leur utilité en toxicopharmacologie au sens large, ces cellules, après différenciation dans le type cellulaire concerné, permettront d’étudier et de modéliser les mécanismes impliqués dans les maladies in vitro mais aussi, par le biais du criblage à haut débit, de mettre au point de nouvelles drogues spécifiques des maladies ciblées.
Les obstacles à l’utilisation des iPS en médecine régénérative
Mais cet énorme potentiel des cellules iPS doit faire face à de nombreux obstacles, résumés ci-dessous, et qui soulèvent à l’heure actuelle la question de leur utilisation en médecine régénérative dans un futur proche [20].
Les vecteurs de transduction. Les plus utilisés sont les rétrovirus et les lentivirus, qui contiennent les séquences codant les différents facteurs responsables de la reprogrammation, inductibles ou non, excisables ou non. Ces vecteurs intégratifs impliquent un risque de mutagenèse insertionnelle et d’activation ou de répression de gènes endogènes [21]. C’est cette approche qui induit la meilleure efficacité de reprogrammation, mais celle-ci reste cependant très faible : de 0,1 % à 0,01 % selon les cellules. Cette efficacité chute de 100 à 1 000 fois lorsque des plasmides, des adénovirus non intégratifs ou des protéines fusion sont utilisés [22].
Les gènes. Tous les gènes de pluripotence utilisés pour produire des iPS sont soit des oncogènes bien établis, Myc et Klf4, soit des protéines liées à la tumorigenèse comme Sox2, Nanog et Oct3/4. Ainsi, il a été récemment montré que c-Myc et Lin28 répriment la transcription de certains microARN, spécialement les membres de la famille LET-7. Cette famille de miARN est souvent réprimée dans les cancers humains, ce qui conduit à la dérépression de leurs cibles comme HMGA-2 (high mobility group), K-Ras and c-Myc et promeut l’oncogenèse [23]. Certains de ces facteurs, en particulier c-Myc et Klf4, ne sont pas indispensables, mais l’efficacité de reprogrammation est dramatiquement réduite en leur absence. La question reste posée du risque de réduction de la « souchitude » des cellules ainsi isolées et/ou, lors de la reprogrammation, de la sélection de cellules exprimant des niveaux élevés de protéines Myc endogènes et potentiellement plus prédisposées à la tumorigenèse [22].
La reprogrammation. Bien que l’extinction des gènes codant les différents facteurs survienne après la reprogrammation, celle-ci est parfois incomplète et une expression résiduelle des facteurs a été mise en évidence. La réactivation peut survenir à des stades plus tardifs comme l’ont montré des études sur les chimères in vivo [24]. Une reprogrammation incomplète ou aberrante peut résulter en la formation de tumeurs malignes après transplantation. La reprogrammation correcte d’une cellule somatique implique que cette dernière subisse des changements épigénétiques (déméthylation de l’ADN, modifications des histones). Une étude récente analysant la méthylation à l’échelon du génome a montré des différences significatives entre le méthylome des cellules iPS et celui des cellules ES [25]. Le moyen de vérifier la pluripotence est la formation de tératomes in vivo. Mais celle-ci ne garantit pas à elle seule la reprogrammation complète des cellules car beaucoup des lignées murines iPS similaires aux cellules ES forment des tératomes mais ne produisent pas de chimères de la lignée germinale. Il est de ce fait difficile de prouver actuellement la reprogrammation complète des cellules humaines [26, 27], et des modèles in vivo de transplantation via différentes voies seront nécessaires pour s’assurer de la réelle pluripotence des cellules. Nul doute que le développement des techniques de criblage à haut débit conduira, dans un futur plus ou moins proche, à l’identification de petites molécules chimiques capables d’induire la reprogrammation [28]. L’alternative plus immédiate sera l’utilisation de systèmes de transfert de gènes non intégratifs et efficaces. Il restera à développer des méthodes permettant d’augmenter l’efficacité de la reprogrammation, à définir des approches pertinentes et les plus stringentes possibles de criblage in vitro et in vivo pour caractériser la pluripotence, l’absence des modifications chromosomiques mineures, et certaines modifications épigénétiques.
Différenciation des iPS en hépatocytes
Nous avons montré que les conditions de différenciation des CSEh en hépatocytes que nous avons décrites permettent d’induire la différenciation d’iPS dérivées de fibroblastes humains en cellules de l’endoderme [29] puis en progéniteurs hépatiques [7]. Ainsi des études de fonctionnalité in vivo pourront être réalisées de façon comparative avec ces deux types de progéniteurs. Nous avons également montré que des iPS générées à partir de patients atteints de maladies métaboliques hépatiques reproduisaient, après différenciation en hépatocytes, le phénotype de la maladie [30].
Autres types de cellules souches sources potentielles d’hépatocytes
Cellules souches d’origine extrahépatique
De nombreux travaux montrent désormais que la transdifférenciation des cellules souches hématopoïétiques en hépatocytes est inefficace à moins que, dans certains modèles de rongeurs, les animaux ne subissent une irradiation (sub)létale préalable, inenvisageable chez l’homme. Dans un certain nombre de cas, ce phénomène correspond à une fusion de cellules hématopoïétiques avec les hépatocytes endogènes [31, 32].
Les cellules souches mésenchymateuses (CSM) sont d’un intérêt potentiel certain, mais les données actuelles suggèrent qu’elles jouent un rôle essentiellement de support trophique dans les foies lésés en inhibant la mort des hépatocytes et en stimulant leur prolifération [33-36]. Notons toutefois que le traitement des CSM dérivées du tissu adipeux et cultivées dans des conditions adaptées aux hépatocytes, par la 5-azacytidine, un inhibiteur puissant des méthyltransférases connu pour induire de façon non spécifique la réactivation de gènes silencieux, induit une différenciation en cellules « de type hépatocyte ». Ces cellules prolifèrent après leur transplantation dans un modèle de souris traitées à la monocrotaline (un alcaloïde à structure pyrrolizidine qui bloque la prolifération des hépatocytes endogènes) et hépatectomisées [37]. Toutefois les CSM, comme toutes les cellules somatiques, ont une capacité d’autorenouvellement limitée, n’ayant pas d’activité télomérase ; en culture, elles entrent en phase de sénescence réplicative après un certain nombre de doublements de population [38, 39].
Cellules souches d’origine hépatique
Les cellules ovales du foie adulte : ce sont des cellules appelées facultative stem cells par nos collègues anglo-saxons car après hépatectomie partielle, seuls les hépatocytes prolifèrent et régénèrent le foie. Chez les rongeurs, la prolifération des cellules ovales est induite dans les situations où les hépatocytes sont détruits par des agents toxiques ou des carcinogènes [40]. Elles sont bipotentes et expriment des marqueurs d’hépatocytes et de cholangiocytes fœtaux comme le font les hépatoblastes. Chez l’homme, après de nombreuses années de controverse, des cellules souches et progénitrices hépatiques ont été isolées non seulement du foie fœtal mais aussi du foie pédiatrique sur la base d’un tri de cellules EpCam+ [12, 41]. Toutefois le phénotype des cellules isolées n’est pas encore complètement défini. De plus, l’activité télomérase mise en évidence dans les foies fœtaux est inexistante dans les cellules souches dérivées des foies nouveau-nés [42].
En conclusion
Dans la mesure où plusieurs milliards d’hépatocytes sont transplantés chez des patients (adultes ou enfants), et même si l’on considère que la transplantation de cellules progénitrices permettra une certaine prolifération in vivo des cellules transplantées avant leur maturation, il sera nécessaire de pouvoir amplifier significativement les différents types de cellules souches avant d’envisager une application en médecine régénérative.
Conflit d’intérêts
Les auteurs déclarent n’avoir aucun conflit d’intérêts concernant les données publiées dans cet article.
Remerciements
Anne Weber et Thomas Touboul souhaitent rendre hommage à la mémoire de Dominique Mahieu-Caputo (chef du service de gynécologie-obstétrique de l’hôpital Bichat) qui a initié et animé le travail sur les hépatoblastes fœtaux humains dans l’unité. L’étude de la différenciation des CSEh en hépatocytes a été financée par l’ANR-05-PRIB-006-02, IngeCell (pôle de compétitivité Medicen) et la CE, dans le cadre du FP7 (projet LIV-ES). Thomas Touboul a été financé par l’AFEF et l’ANR-RIB.
L’immunodéficience de ces souris s’explique par l’absence du gène Rag, codant l’enzyme responsable de la recombinaison génétique des gènes codant les immunoglobulines indispensable à la formation des récepteurs T et B, et par l’absence du gène codant la chaîne γ du récepteur de cytokines, ce qui bloque la prolifération des populations lymphoïdes.
Références
- D’Amour KA, Bang AG, Eliazer S, et al. Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells. Nat Biotechnol 2006 ; 24 : 1392-401. [Google Scholar]
- Hay DC, Fletcher J, Payne C, et al. Highly efficient differentiation of hESCs to functional hepatic endoderm requires ActivinA and Wnt3a signaling. Proc Natl Acad Sci USA 2008 ; 105 : 12301-6. [Google Scholar]
- Cai J, Zhao Y, Liu Y, et al. Directed differentiation of human embryonic stem cells into functional hepatic cells. Hepatology 2007 ; 45 : 1229-39. [Google Scholar]
- Si-Tayeb K, Noto FK, Nagaoka M, et al. Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology 2010 ; 51 : 297-305. [Google Scholar]
- Hay DC, Zhao D, Fletcher J, et al. Efficient differentiation of hepatocytes from human embryonic stem cells exhibiting markers recapitulating liver development in vivo. Stem Cells 2008 ; 26 : 894-902. [Google Scholar]
- Basma H, Soto-Gutierrez A, Yannam GR, et al. Differentiation and transplantation of human embryonic stem cell-derived hepatocytes. Gastroenterology 2009 ; 136 : 990-9. [Google Scholar]
- Touboul T, Hannan NR, Corbineau S, et al. Generation of functional hepatocytes from human embryonic stem cells under chemically defined conditions that recapitulate liver development. Hepatology 2010 (sous presse). [Google Scholar]
- McLean AB, D’Amour KA, Jones KL, et al. Activin a efficiently specifies definitive endoderm from human embryonic stem cells only when phosphatidylinositol 3-kinase signaling is suppressed. Stem Cells 2007 ; 25 : 29-38. [Google Scholar]
- Vallier L, Touboul T, Chng Z, et al. Early cell fate decisions of human embryonic stem cells and mouse epiblast stem cells are controlled by the same signalling pathways. PLoS One 2009 ; : e6082. [Google Scholar]
- Zaret KS. Genetic programming of liver and pancreas progenitors : lessons for stem-cell differentiation. Nat Rev Genet 2008 ; 9 : 329-40. [Google Scholar]
- Berg T, Rountree CB, Lee L, et al. Fibroblast growth factor 10 is critical for liver growth during embryogenesis and controls hepatoblast survival via beta-catenin activation. Hepatology 2007 ; 46 : 1187-97. [Google Scholar]
- Schmelzer E, Zhang L, Bruce A, et al. Human hepatic stem cells from fetal and postnatal donors. J Exp Med 2007 ; 204 : 1973-87. [Google Scholar]
- Odom DT, Zizlsperger N, Gordon DB, et al. Control of pancreas and liver gene expression by HNF transcription factors. Science 2004 ; 303 : 1378-81. [Google Scholar]
- Bachoud-Levi AC, Gaura V, Brugieres P, et al. Effect of fetal neural transplants in patients with Huntington’s disease 6 years after surgery : a long-term follow-up study. Lancet Neurol 2006 ; 5 : 303-9. [Google Scholar]
- Mahieu-Caputo D, Allain JE, Branger J, et al. Repopulation of athymic mouse liver by cryopreserved early human fetal hepatoblasts. Hum Gene Ther 2004 ; 15 : 1219-28. [Google Scholar]
- Carr AJ, Vugler AA, Hikita ST, et al. Protective effects of human iPS-derived retinal pigment epithelium cell transplantation in the retinal dystrophic rat. PLoS One 2009 ; 4 : e8152. [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006 ; 126 : 663-76. [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007 ; 131 : 861-72. [Google Scholar]
- Aoi T, Yae K, Nakagawa M, et al. Generation of pluripotent stem cells from adult mouse liver and stomach cells. Science 2008 ; 321 : 699-702. [Google Scholar]
- Robbins RD, Prasain N, Maier BF, et al. Inducible pluripotent stem cells : not quite ready for prime time? Curr Opin Organ Transplant 2010 ; 15 : 61-7. [Google Scholar]
- Montini E, Cesana D, Schmidt M, et al. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. J Clin Invest 2009 ; 119 : 964-75. [Google Scholar]
- Jalving M, Shepers H. Induced pluripotent stem cells : will they be safe? Curr Opin Mol Ther 2009 ; 11 : 383-93. [Google Scholar]
- Viswanathan SR, Powers JT, Einhorn W, et al. Lin28 promotes transformation and is associated with advanced human malignancies. Nat Genet 2009 ; 41 : 843-8. [Google Scholar]
- Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature 2007 ; 448 : 313-7. [Google Scholar]
- Doi A, Park IH, Wen B, et al. Differential methylation of tissue- and cancerspecific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat Genet 2009 ; 41 : 1350-3. [Google Scholar]
- Yamanaka S. A fresh look at iPS cells. Cell 2009 ; 137 : 13-7. [Google Scholar]
- Coulombel L. Pluripotence : une définition à géométrie variable. Med Sci (Paris) 2009 ; 25 : 798-801. [Google Scholar]
- Lin T, Ambasudhan R, Yuan X, et al. A chemical platform for improved induction of human iPSCs. Nat Methods 2009 ; 6 : 805-8. [Google Scholar]
- Vallier L, Touboul T, Brown S, et al. Signaling pathways controlling pluripotency and early cell fate decisions of human induced pluripotent stem cells. Stem Cells 2009 ; 27 : 2655-66. [Google Scholar]
- Rashid ST, Corbineau S, Hannan N, et al. Modeling inherited metabolic disorders of the liver using human induced pluripotent stem cells. J Clin Invest 2010 ; 120 : 3127-36. [Google Scholar]
- Thorgeirsson SS, Grisham JW. Hematopoietic cells as hepatocyte stem cells : a critical review of the evidence. Hepatology 2006 ; 43 : 2-8. [Google Scholar]
- Fujino H, Hiramatsu H, Tsuchiya A, et al. Human cord blood CD34+ cells develop into hepatocytes in the livers of NOD/SCID/gamma(c)null mice through cell fusion. FASEB J 2007 ; 21 : 3499-510. [Google Scholar]
- van Poll D, Parekkadan B, Cho CH, et al. Mesenchymal stem cell-derived molecules directly modulate hepatocellular death and regeneration in vitro and in vivo. Hepatology 2008 ; 47 : 1634-43. [Google Scholar]
- Banas A, Teratani T, Yamamoto Y, et al. Adipose tissue-derived mesenchymal stem cells as a source of human hepatocytes. Hepatology 2007 ; 46 : 219-28. [Google Scholar]
- Tsai PC, Fu TW, Chen YM, et al. The therapeutic potential of human umbilical mesenchymal stem cells from Wharton’s jelly in the treatment of rat liver fibrosis. Liver Transpl 2009 ; 15 : 484-95. [Google Scholar]
- Yan Y, Xu W, Qian H, et al. Mesenchymal stem cells from human umbilical cords ameliorate mouse hepatic injury in vivo. Liver Int 2009 ; 29 : 356-65. [Google Scholar]
- Banas A, Teratani T, Yamamoto Y, et al. Rapid hepatic fate specification of adipose-derived stem cells and their therapeutic potential for liver failure. J Gastroenterol Hepatol 2009 ; 24 : 70-7. [Google Scholar]
- Bernardo ME, Zaffaroni N, Novara F, et al. Human bone marrow derived mesenchymal stem cells do not undergo transformation after long-term in vitro culture and do not exhibit telomere maintenance mechanisms. Cancer Res 2007 ; 67 : 9142-9. [Google Scholar]
- Wagner W, Bork S, Horn P, et al. Aging and replicative senescence have related effects on human stem and progenitor cells. PLoS One 2009 ; 4 : e5846. [Google Scholar]
- Wang X, Foster M, Al-Dhalimy M, et al. The origin and liver repopulating capacity of murine oval cells. Proc Natl Acad Sci USA 2003 ; 100 (suppl 1) : 11881-8. [Google Scholar]
- Wauthier E, Schmelzer E, Turner W, et al. Hepatic stem cells and hepatoblasts : identification, isolation, and ex vivo maintenance. Methods Cell Biol 2008 ; 86 : 137-225. [Google Scholar]
- Schmelzer E, Reid LM. Human telomerase activity, telomerase and telomeric template expression in hepatic stem cells and in livers from fetal and postnatal donors. Eur J Gastroenterol Hepatol 2009 ; 21 : 1191-8. [Google Scholar]
- Gires O. EpCAM, une protéine versatile impliquée dans l’oncogenèse. Med Sci (Paris) 2009 ; 25 : 449-50. [Google Scholar]
- Coulombel L. Coup de coeur pour un progéniteur multipotent mésodermique dérivé de cellules souches embryonnaires humaines. Med Sci (Paris) 2010 ; 26 : 439-41. [Google Scholar]
- Coulombel L. Reprogrammation nucléaire d’une cellule différenciée : on efface tout et on recommence. Med Sci (Paris) 2007 ; 23 : 667-70. [Google Scholar]
- Zorn AM, Wells JM. Vertebrate endoderm development and organ formation. Annu Rev Cell Dev Biol 2009 ; 25 : 221-51. [Google Scholar]
Liste des figures
|
Figure 1 Origine et formation de l’endoderme. À gauche : résumé des événements se produisant lors de la formation de l’endoderme qui s’accompagne d’un changement de morphologie de l’embryon de souris. L’endoderme est représenté en jaune. L’endoderme et le mésoderme dériveraient d’un progéniteur commun, le mésendoderme. À droite : différents lignages dérivés de l’endoderme projetés sur une représentation du tractus gastrointestinal (d’après [46]). |
| Dans le texte | |
|
Figure 2 Différenciation des CSEh en hépatocytes. Les CSEh indifférenciées progressent dans la voie hépatique par étapes successives correspondant à l’expression transitoire de certains marqueurs comme Sox17 et CXCR4 au stade de l’endoderme définitif (DE), EpCam et N-Cam au stade des cellules souches hépatiques (hHpSC). Au cours de leur différenciation, les cellules changent de morphologie pour adopter, au stade hépatocytes, une forme pentagonale caracté- ristique. FC : fetal cholangiocytes ; FH : fetal hepatocytes. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.